

Animal models of muscular dystrophy
- PMID: 22137430
- PMCID: PMC4872622
- DOI: 10.1016/B978-0-12-394596-9.00004-4
Animal models of muscular dystrophy
Abstract
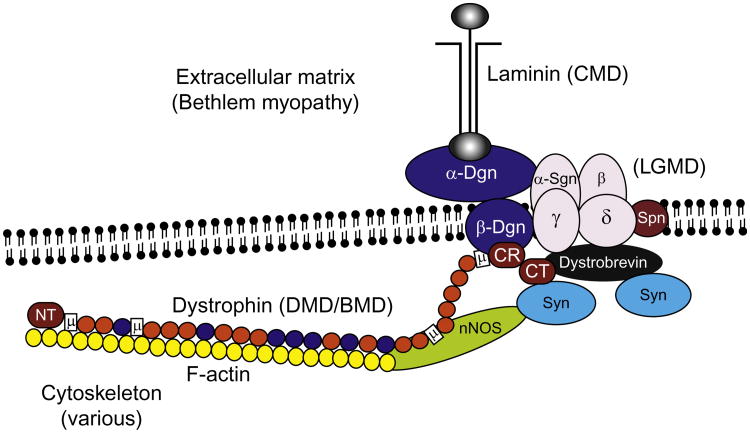
The muscular dystrophies (MDs) represent a diverse collection of inherited human disorders, which affect to varying degrees skeletal, cardiac, and sometimes smooth muscle (Emery, 2002). To date, more than 50 different genes have been implicated as causing one or more types of MD (Bansal et al., 2003). In many cases, invaluable insights into disease mechanisms, structure and function of gene products, and approaches for therapeutic interventions have benefited from the study of animal models of the different MDs (Arnett et al., 2009). The large number of genes that are associated with MD and the tremendous number of animal models that have been developed preclude a complete discussion of each in the context of this review. However, we summarize here a number of the more commonly used models together with a mixture of different types of gene and MD, which serves to give a general overview of the value of animal models of MD for research and therapeutic development.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Emery AE. The muscular dystrophies. Lancet. 2002;359:687–95. - PubMed
-
- Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–72. - PubMed
-
- Arnett AL, Chamberlain JR, Chamberlain JS. Therapy for neuromuscular disorders. Curr Opin Genet Dev. 2009;19:290–7. - PubMed
-
- Kunkel LM, et al. Analysis of deletions in DNA from patients with Becker and Duchenne muscular dystrophy. Nature. 1986;322:73–7. - PubMed
-
- Monaco AP, Neve RL, Coletti-Feener C, Bertelson CJ, Kurnit DM, Kunkel LM. Isolation of candidate cDNA clones for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323:646–50. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
