

CFTR: folding, misfolding and correcting the ΔF508 conformational defect
- PMID: 22138491
- PMCID: PMC3643519
- DOI: 10.1016/j.molmed.2011.10.003
CFTR: folding, misfolding and correcting the ΔF508 conformational defect
Abstract
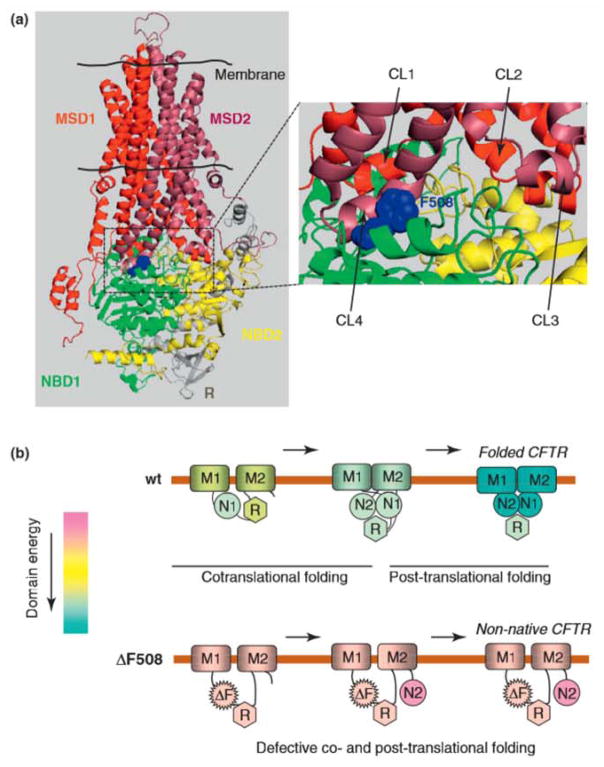
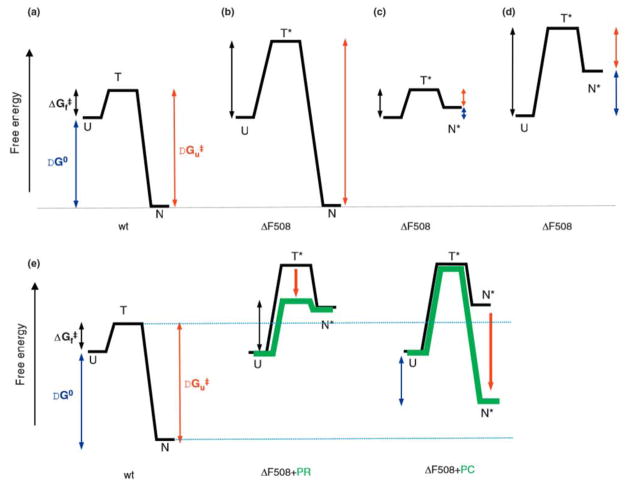
Cystic fibrosis (CF), the most common lethal genetic disease in the Caucasian population, is caused by loss-of-function mutations of the CF transmembrane conductance regulator (CFTR), a cyclic AMP-regulated plasma membrane chloride channel. The most common mutation, deletion of phenylalanine 508 (ΔF508), impairs CFTR folding and, consequently, its biosynthetic and endocytic processing as well as chloride channel function. Pharmacological treatments may target the ΔF508 CFTR structural defect directly by binding to the mutant protein and/or indirectly by altering cellular protein homeostasis (proteostasis) to promote ΔF508 CFTR plasma membrane targeting and stability. This review discusses recent basic research aimed at elucidating the structural and trafficking defects of ΔF508 CFTR, a prerequisite for the rational design of CF therapy to correct the loss-of-function phenotype.
Crown Copyright © 2011. Published by Elsevier Ltd. All rights reserved.
Figures



References
-
- Riordan JR. Assembly or functional CFTR chloride channels. Annu Rev Physiol. 2005;67:701–718. - PubMed
-
- Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–726. - PubMed
-
- Cheng SH, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. - PubMed
-
- Qu BH, Thomas PJ. Alteration of the cystic fibrosis transmembrane conductance regulator folding pathway. J Biol Chem. 1996;271:7261–7264. - PubMed
-
- Lukacs GL, et al. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem. 1993;268:21592–21598. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
