

New understandings of the genetic basis of isolated idiopathic central hypogonadism
- PMID: 22138902
- PMCID: PMC3735150
- DOI: 10.1038/aja.2011.68
New understandings of the genetic basis of isolated idiopathic central hypogonadism
Abstract
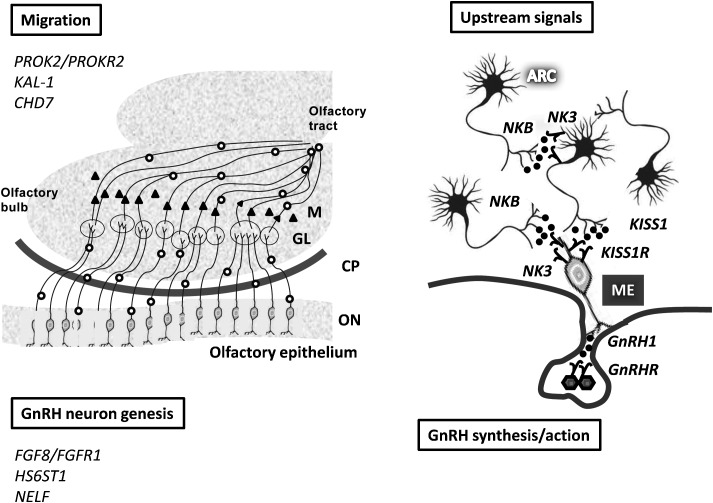
Idiopathic hypogonadotropic hypogonadism is a rare disease that is characterized by delayed/absent puberty and/or infertility due to an insufficient stimulation of an otherwise normal pituitary-gonadal axis by gonadotrophin-releasing hormone (GnRH) action. Because reduced or normal luteinizing hormone (LH)/follicle-stimulating hormone (FSH) levels may be observed in the affected patients, the term idiopathic central hypogonadism (ICH) appears to be more appropriate. This disease should be distinguished from central hypogonadism that is combined with other pituitary deficiencies. Isolated ICH has a complex pathogenesis and is fivefold more prevalent in males. ICH frequently appears in a sporadic form, but several familial cases have also been reported. This finding, in conjunction with the description of numerous pathogenetic gene variants and the generation of several knockout models, supports the existence of a strong genetic component. ICH may be associated with several morphogenetic abnormalities, which include osmic defects that, with ICH, constitute the cardinal manifestations of Kallmann syndrome (KS). KS accounts for approximately 40% of the total ICH cases and has been generally considered to be a distinct subgroup. However, the description of several pedigrees, which include relatives who are affected either with isolated osmic defects, KS, or normo-osmic ICH (nICH), justifies the emerging idea that ICH is a complex genetic disease that is characterized by variable expressivity and penetrance. In this context, either multiple gene variants or environmental factors and epigenetic modifications may contribute to the variable disease manifestations. We review the genetic mechanisms that are presently known to be involved in ICH pathogenesis and provide a clinical overview of the 227 cases that have been collected by the collaborating centres of the Italian ICH Network.
Figures
References
-
- Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. 1989;338:161–4. - PubMed
-
- Gonzalez-Martinez D, Hu Y, Bouloux PM. Ontogeny of GnRH and olfactory neuronal systems in man: novel insights from the investigation of inherited forms of Kallmann's syndrome. Front Neuroendocrinol. 2004;25:108–30. - PubMed
-
- Cadman SM, Kim SH, Hu Y, Gonzalez-Martinez D, Bouloux PM. Molecular pathogenesis of Kallmann's syndrome. Horm Res. 2007;67:231–42. - PubMed
-
- Mitchell AL, Dwyer A, Pitteloud N, Quinton R. Genetic basis and variable phenotypic expression of Kallmann syndrome: towards a unifying theory. Trends Endocrinol Metab. 2011;22:249–58. - PubMed
