

Sympathetic nerve stimulation induces local endothelial Ca2+ signals to oppose vasoconstriction of mouse mesenteric arteries
- PMID: 22140050
- PMCID: PMC3353782
- DOI: 10.1152/ajpheart.00773.2011
Sympathetic nerve stimulation induces local endothelial Ca2+ signals to oppose vasoconstriction of mouse mesenteric arteries
Abstract
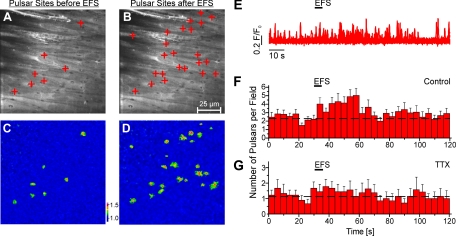
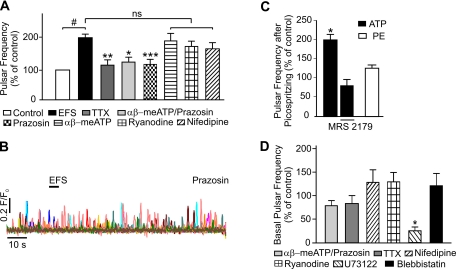
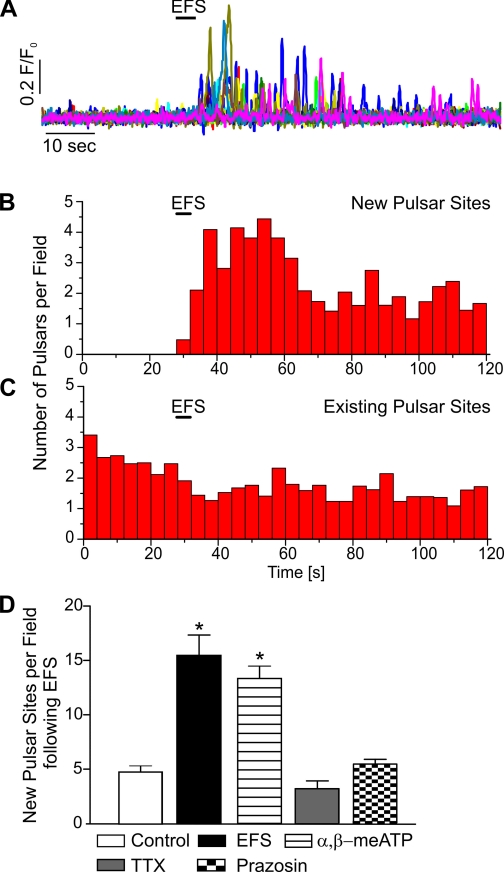
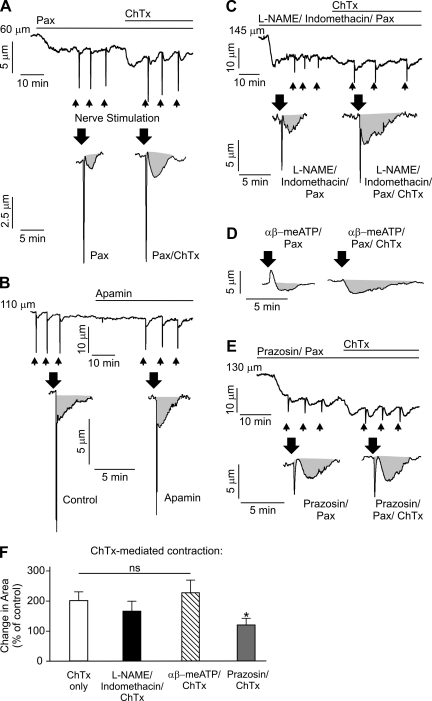
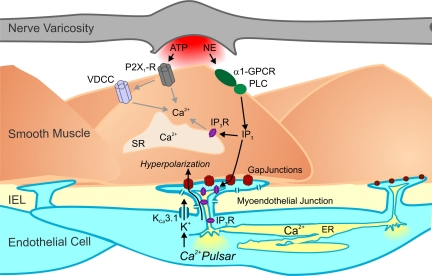
It is generally accepted that the endothelium regulates vascular tone independent of the activity of the sympathetic nervous system. Here, we tested the hypothesis that the activation of sympathetic nerves engages the endothelium to oppose vasoconstriction. Local inositol 1,4,5-trisphosphate (IP(3))-mediated Ca(2+) signals ("pulsars") in or near endothelial projections to vascular smooth muscle (VSM) were measured in an en face mouse mesenteric artery preparation. Electrical field stimulation of sympathetic nerves induced an increase in endothelial cell (EC) Ca(2+) pulsars, recruiting new pulsar sites without affecting activity at existing sites. This increase in Ca(2+) pulsars was blocked by bath application of the α-adrenergic receptor antagonist prazosin or by TTX but was unaffected by directly picospritzing the α-adrenergic receptor agonist phenylephrine onto the vascular endothelium, indicating that nerve-derived norepinephrine acted through α-adrenergic receptors on smooth muscle cells. Moreover, EC Ca(2+) signaling was not blocked by inhibitors of purinergic receptors, ryanodine receptors, or voltage-dependent Ca(2+) channels, suggesting a role for IP(3), rather than Ca(2+), in VSM-to-endothelium communication. Block of intermediate-conductance Ca(2+)-sensitive K(+) channels, which have been shown to colocalize with IP(3) receptors in endothelial projections to VSM, enhanced nerve-evoked constriction. Collectively, our results support the concept of a transcellular negative feedback module whereby sympathetic nerve stimulation elevates EC Ca(2+) signals to oppose vasoconstriction.
Figures
References
-
- Allbritton NL, Meyer T, Stryer L. Range of messenger action of calcium ion and inositol 1,4,5-trisphosphate. Science 258: 1812–1815, 1992 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous