

Regulation of alveolar epithelial cell apoptosis and pulmonary fibrosis by coordinate expression of components of the fibrinolytic system
- PMID: 22140072
- PMCID: PMC3311514
- DOI: 10.1152/ajplung.00099.2011
Regulation of alveolar epithelial cell apoptosis and pulmonary fibrosis by coordinate expression of components of the fibrinolytic system
Abstract
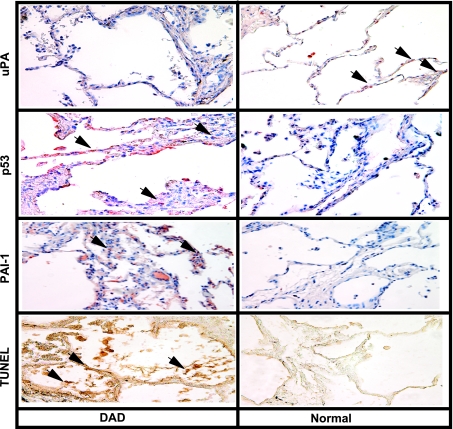
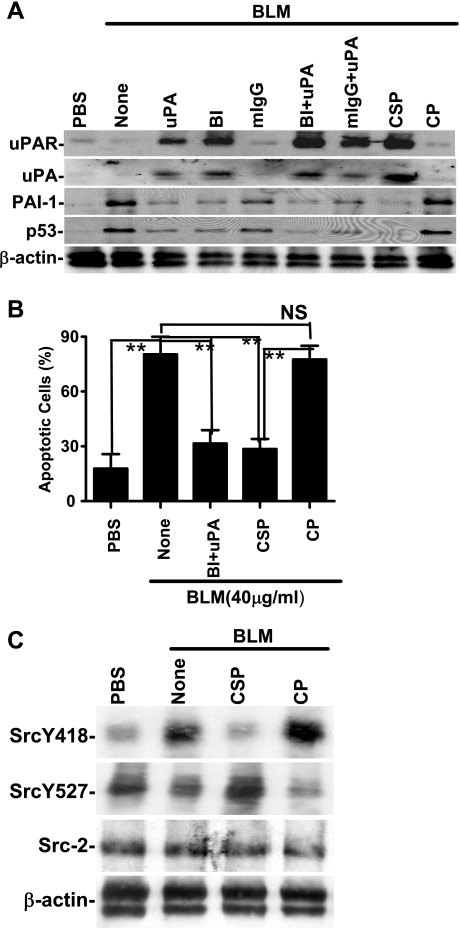
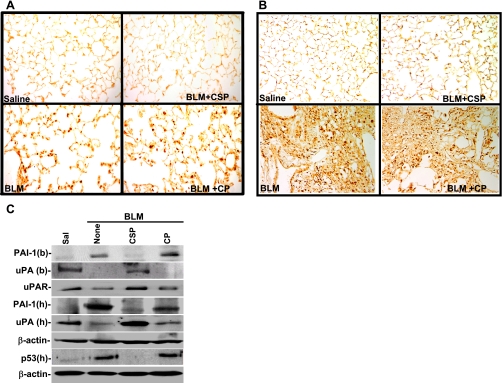
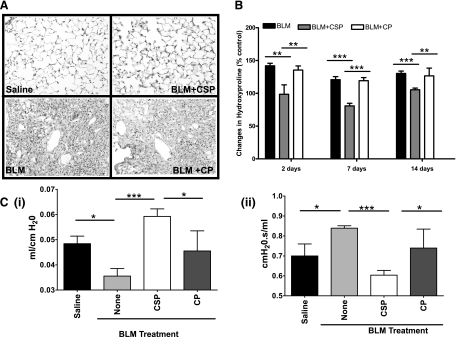
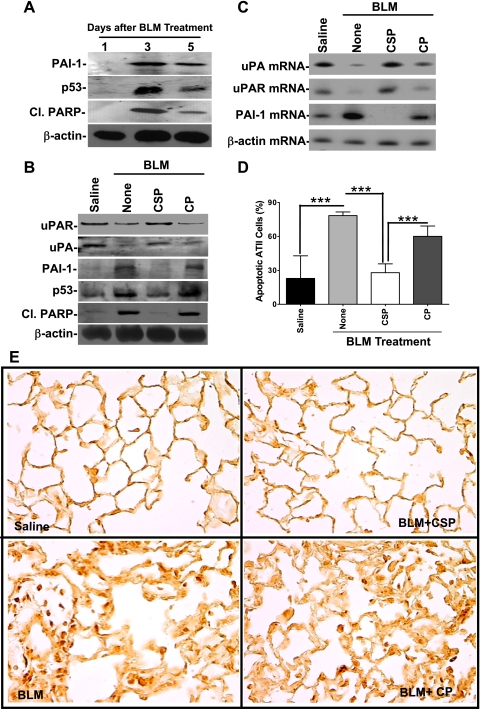
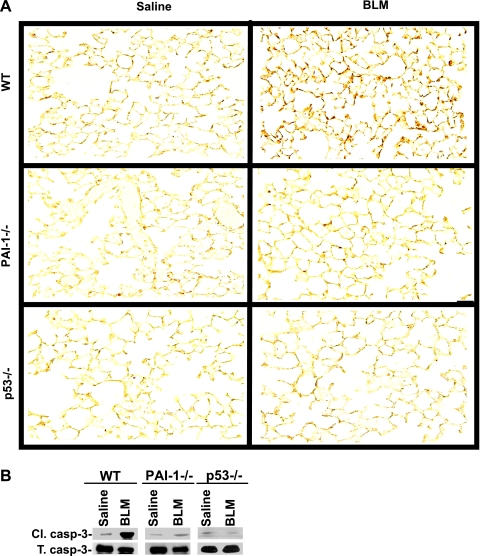
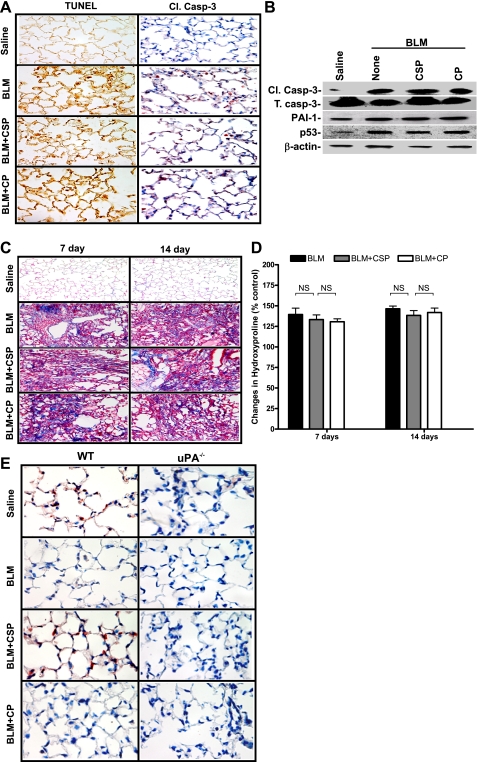
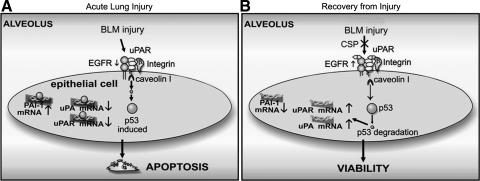
Alveolar type II (ATII) cell apoptosis and depressed fibrinolysis that promotes alveolar fibrin deposition are associated with acute lung injury (ALI) and the development of pulmonary fibrosis (PF). We therefore sought to determine whether p53-mediated inhibition of urokinase-type plasminogen activator (uPA) and induction of plasminogen activator inhibitor-1 (PAI-1) contribute to ATII cell apoptosis that precedes the development of PF. We also sought to determine whether caveolin-1 scaffolding domain peptide (CSP) reverses these changes to protect against ALI and PF. Tissues as well as isolated ATII cells from the lungs of wild-type (WT) mice with BLM injury show increased apoptosis, p53, and PAI-1, and reciprocal suppression of uPA and uPA receptor (uPAR) protein expression. Treatment of WT mice with CSP reverses these effects and protects ATII cells against bleomycin (BLM)-induced apoptosis whereas CSP fails to attenuate ATII cell apoptosis or decrease p53 or PAI-1 in uPA-deficient mice. These mice demonstrate more severe PF. Thus p53 is increased and inhibits expression of uPA and uPAR while increasing PAI-1, changes that promote ATII cell apoptosis in mice with BLM-induced ALI. We show that CSP, an intervention targeting this pathway, protects the lung epithelium from apoptosis and prevents PF in BLM-induced lung injury via uPA-mediated inhibition of p53 and PAI-1.
Figures
References
-
- Alfano D, Franco P, Vocca I, Gambi N, Pisa V, Mancini A, Caputi M, Carriero MV, Iaccarino I, Stoppelli MP. The urokinase plasminogen activator and its receptor: role in cell growth and apoptosis. Thromb Haemost 93: 205– 211, 2005 - PubMed
-
- Barker SC, Kassel DB, Weigl D, Huang X, Luther MA, Knight WB. Characterization of pp60c-src tyrosine kinase activities using a continuous assay: autoactivation of the enzyme is an intermolecular autophosphorylation process. Biochemistry 34: 14843– 14851, 1995 - PubMed
-
- Bertozzi P, Astedt B, Zenzius L, Lynch K, LeMaire F, Zapol W, Chapman HA., Jr Depressed bronchoalveolar urokinase activity in patients with adult respiratory distress syndrome. N Engl J Med 322: 890– 897, 1990 - PubMed
-
- Bhandary YP, Shetty SK, Shetty R, Idell S, Starcher B, Shetty S. Regulation of lung epithelial apoptosis by coordinate expression of components of the fibrinolytic system (Abstract). Am J Respir Crit Care Med 181: A4185, 2010
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous