

Miscellaneous non-inflammatory musculoskeletal conditions. Hyperphosphatemic familial tumoral calcinosis (FGF23, GALNT3 and αKlotho)
- PMID: 22142751
- PMCID: PMC3233725
- DOI: 10.1016/j.berh.2011.10.020
Miscellaneous non-inflammatory musculoskeletal conditions. Hyperphosphatemic familial tumoral calcinosis (FGF23, GALNT3 and αKlotho)
Abstract
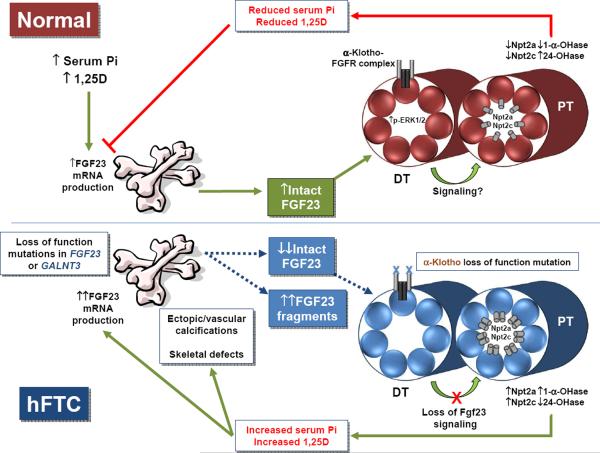
Familial tumoral calcinosis (TC) is a rare disorder distinguished by the development of ectopic and vascular calcified masses that occur in settings of hyperphosphatemia (hFTC) and normophosphatemia (nFTC). Serum phosphorus concentrations are relatively tightly controlled by interconnected endocrine activity at the level of the intestine, kidney, and skeleton. Discovering the molecular causes for heritable forms of hFTC has shed new light on the regulation of serum phosphate balance. This review will focus upon the genetic basis and clinical approaches for hFTC, due to genes that are related to the phosphaturic hormone fibroblast growth factor-23 (FGF23). These include FGF23 itself, an FGF23-glycosylating enzyme (GALNT3), and the FGF23 co-receptor α-Klotho (αKL). Our understanding of the molecular basis of hFTC will, in the short term, aid in understanding normal phosphate balance, and in the future, provide potential insight into the design of novel therapeutic strategies for both rare and common disorders of phosphate metabolism.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Familial tumoral calcinosis and the role of O-glycosylation in the maintenance of phosphate homeostasis.Biochim Biophys Acta. 2009 Sep;1792(9):847-52. doi: 10.1016/j.bbadis.2008.10.008. Epub 2008 Oct 25. Biochim Biophys Acta. 2009. PMID: 19013236 Free PMC article. Review.
-
Hyperphosphatemic familial tumoral calcinosis: genetic models of deficient FGF23 action.Curr Osteoporos Rep. 2015 Apr;13(2):78-87. doi: 10.1007/s11914-015-0254-3. Curr Osteoporos Rep. 2015. PMID: 25656441 Review.
-
The role of mutant UDP-N-acetyl-alpha-D-galactosamine-polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis.J Clin Endocrinol Metab. 2006 Oct;91(10):4037-42. doi: 10.1210/jc.2006-0305. Epub 2006 Jul 25. J Clin Endocrinol Metab. 2006. PMID: 16868048
-
Ablation of the Galnt3 gene leads to low-circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression.Endocrinology. 2009 Jun;150(6):2543-50. doi: 10.1210/en.2008-0877. Epub 2009 Feb 12. Endocrinology. 2009. PMID: 19213845 Free PMC article.
-
Phenotypic and Genotypic Characterization and Treatment of a Cohort With Familial Tumoral Calcinosis/Hyperostosis-Hyperphosphatemia Syndrome.J Bone Miner Res. 2016 Oct;31(10):1845-1854. doi: 10.1002/jbmr.2870. Epub 2016 Sep 20. J Bone Miner Res. 2016. PMID: 27164190 Free PMC article. Clinical Trial.
Cited by
-
Recurrent Bilateral Lower Motor Neuron Type of Facial Palsy with Hearing Impairment: Hyperphosphatemic Familial Tumoral Calcinosis.J Pediatr Genet. 2022 Jan 7;12(4):280-287. doi: 10.1055/s-0041-1741522. eCollection 2023 Dec. J Pediatr Genet. 2022. PMID: 38162162 Free PMC article.
-
Pseudoxanthoma Elasticum as a Paradigm of Heritable Ectopic Mineralization Disorders: Pathomechanisms and Treatment Development.Am J Pathol. 2019 Feb;189(2):216-225. doi: 10.1016/j.ajpath.2018.09.014. Epub 2018 Nov 7. Am J Pathol. 2019. PMID: 30414410 Free PMC article. Review.
-
Hyperphosphatemic Familial Tumoral Calcinosis With Galnt3 Mutation: Transient Response to Anti-Interleukin-1 Treatments.JBMR Plus. 2019 Mar 6;3(7):e10185. doi: 10.1002/jbm4.10185. eCollection 2019 Jul. JBMR Plus. 2019. PMID: 31372591 Free PMC article.
-
Identification of two novel mutations in the GALNT3 gene in a Chinese family with hyperphosphatemic familial tumoral calcinosis.Bone Res. 2016 Nov 8;4:16038. doi: 10.1038/boneres.2016.38. eCollection 2016. Bone Res. 2016. PMID: 27867679 Free PMC article.
-
A Case of Hyperphosphatemia and Elevated Fibroblast Growth Factor 23: A Brief Review of Hyperphosphatemia and Fibroblast Growth Factor 23 Pathway.Kidney Int Rep. 2017 May 17;2(6):1238-1242. doi: 10.1016/j.ekir.2017.05.006. eCollection 2017 Nov. Kidney Int Rep. 2017. PMID: 29270533 Free PMC article. No abstract available.
References
-
- Berndt TJ, Knox FG. In: The Kidney: Physiology and Pathophysiology. Seldin DW, Giebisch G, editors. Raven; New York: 1992. pp. 2511–2532.
-
- Gmaj P, Murer H. Cellular mechanisms of inorganic phosphate transport in kidney. Physiol Rev. 1986;66:36–70. - PubMed
-
- Mizgala CL, Quamme GA. Renal handling of phosphate. Physiol Rev. 1985;65:431–466. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
