

Myopathy caused by mammalian target of rapamycin complex 1 (mTORC1) inactivation is not reversed by restoring mitochondrial function
- PMID: 22143799
- PMCID: PMC3251091
- DOI: 10.1073/pnas.1111448109
Myopathy caused by mammalian target of rapamycin complex 1 (mTORC1) inactivation is not reversed by restoring mitochondrial function
Abstract
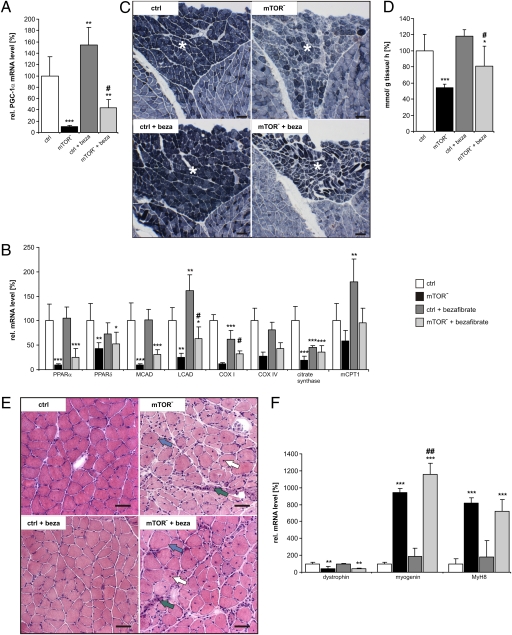
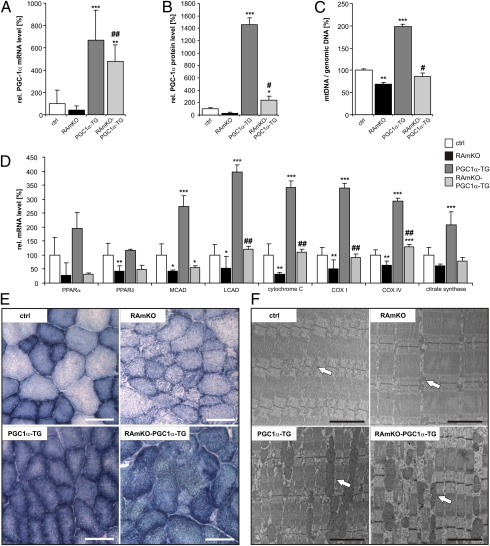
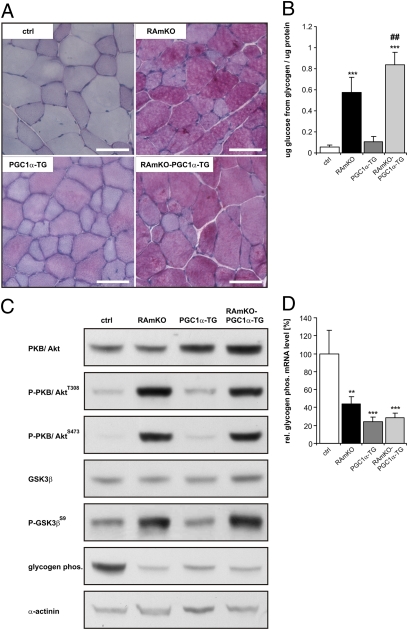
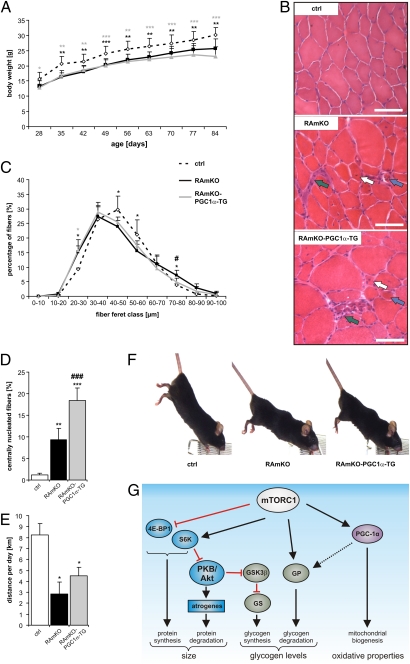
Mammalian target of rapamycin complex 1 (mTORC1) is central to the control of cell, organ, and body size. Skeletal muscle-specific inactivation of mTORC1 in mice results in smaller muscle fibers, fewer mitochondria, increased glycogen stores, and a progressive myopathy that causes premature death. In mTORC1-deficient muscles, peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α), which regulates mitochondrial biogenesis and glucose homeostasis, is strongly down-regulated. Here we tested whether induction of mitochondrial biogenesis pharmacologically or by the overexpression of PGC-1α is sufficient to reverse the phenotype of mice deficient for mTORC1. We show that both approaches normalize mitochondrial function, such as oxidative capacity and expression of mitochondrial genes. However, they do not prevent or delay the progressive myopathy. In addition, we find that mTORC1 has a much stronger effect than PGC-1α on the glycogen content in muscle. This effect is based on the strong activation of PKB/Akt in mTORC1-deficient mice. We also show that activation of PKB/Akt not only affects glycogen synthesis but also diminishes glycogen degradation. Thus, our work provides strong functional evidence that mitochondrial dysfunction in mice with inactivated mTORC1 signaling is caused by the down-regulation of PGC-1α. However, our data also show that the impairment of mitochondria does not lead directly to the lethal myopathy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol. 2005;37:1974–1984. - PubMed
-
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. - PubMed
-
- Bentzinger CF, et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 2008;8:411–424. - PubMed
-
- Um SH, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
