

TAK1 in brain endothelial cells mediates fever and lethargy
- PMID: 22143887
- PMCID: PMC3244031
- DOI: 10.1084/jem.20110398
TAK1 in brain endothelial cells mediates fever and lethargy
Abstract
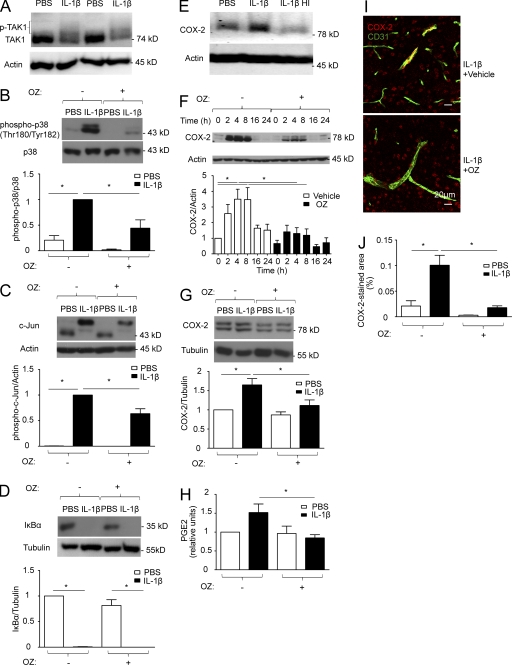
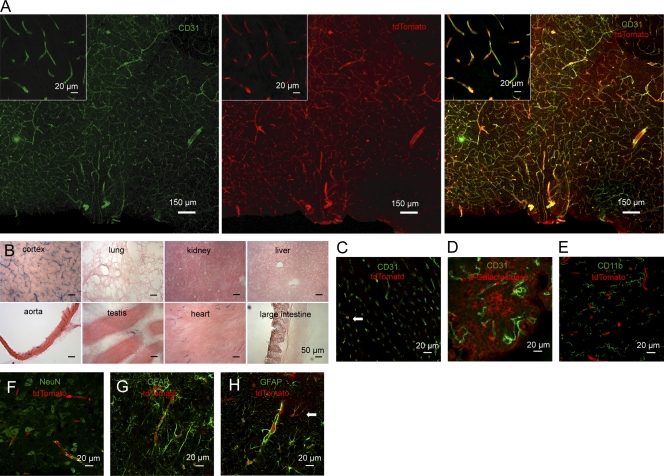
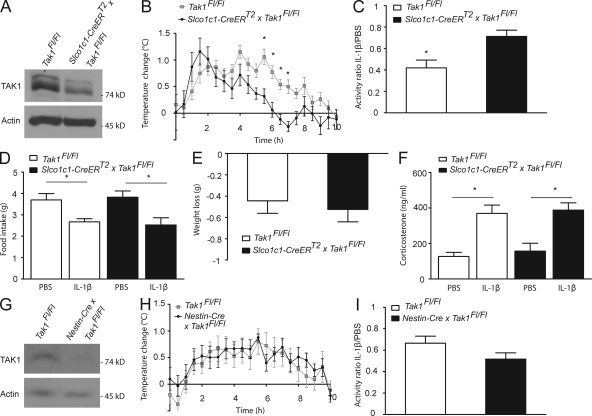
Systemic inflammation affects the brain, resulting in fever, anorexia, lethargy, and activation of the hypothalamus-pituitary-adrenal axis. How peripheral inflammatory signals reach the brain is still a matter of debate. One possibility is that, in response to inflammatory stimuli, brain endothelial cells in proximity to the thermoregulatory centers produce cyclooxygenase 2 (COX-2) and release prostaglandin E2, causing fever and sickness behavior. We show that expression of the MAP kinase kinase kinase TAK1 in brain endothelial cells is needed for interleukin 1β (IL-1β)-induced COX-2 production. Exploiting the selective expression of the thyroxine transporter Slco1c1 in brain endothelial cells, we generated a mouse line allowing inducible deletion of Tak1 specifically in brain endothelium. Mice lacking the Tak1 gene in brain endothelial cells showed a blunted fever response and reduced lethargy upon intravenous injection of the endogenous pyrogen IL-1β. In conclusion, we demonstrate that TAK1 in brain endothelial cells induces COX-2, most likely by activating p38 MAPK and c-Jun, and is necessary for fever and sickness behavior.
Figures
References
-
- Ching S., Zhang H., Belevych N., He L., Lai W., Pu X.A., Jaeger L.B., Chen Q., Quan N. 2007. Endothelial-specific knockdown of interleukin-1 (IL-1) type 1 receptor differentially alters CNS responses to IL-1 depending on its route of administration. J. Neurosci. 27:10476–10486 10.1523/JNEUROSCI.3357-07.2007 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
