

Critical requirement for Stat5 in a mouse model of polycythemia vera
- PMID: 22144185
- PMCID: PMC3325041
- DOI: 10.1182/blood-2011-03-345215
Critical requirement for Stat5 in a mouse model of polycythemia vera
Abstract
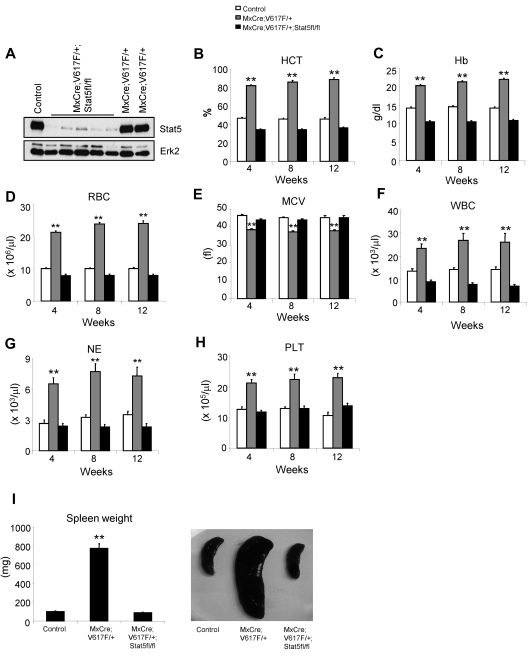
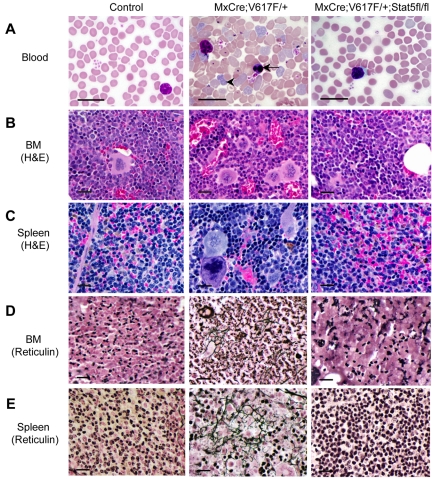
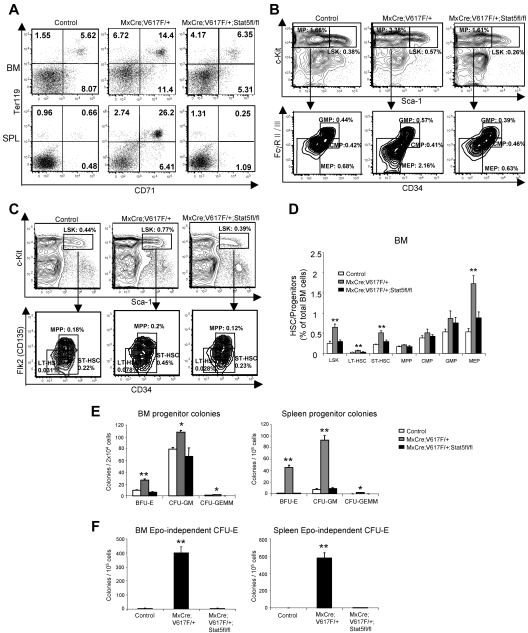
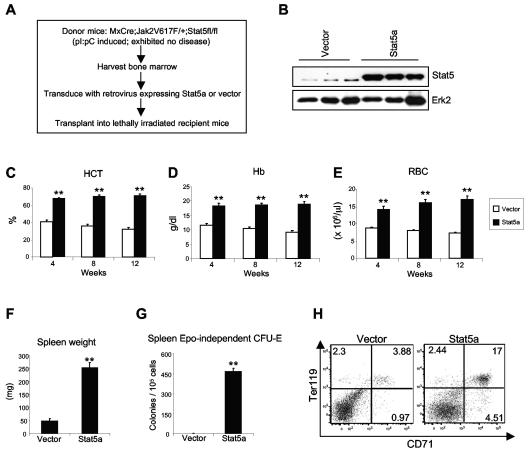
The JAK2V617F mutation has been identified in most cases of Ph-negative myeloproliferative neoplasms (MPNs) including polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). Expression of JAK2V617F results in constitutive activation of multiple signaling molecules/pathways. However, the key signaling downstream of JAK2V617F required for transformation and induction of MPNs remains elusive. Using a mouse genetic strategy, we show here that Stat5 is absolutely required for the pathogenesis of PV induced by Jak2V617F. Whereas expression of Jak2V617F in mice resulted in all the features of human PV, including an increase in red blood cells, hemoglobin, hematocrit, white blood cells, platelets, and splenomegaly, deletion of Stat5 in the Jak2V617F knockin mice normalized all the blood parameters and the spleen size. Furthermore, deletion of Stat5 completely abrogated erythropoietin (Epo)-independent erythroid colony formation evoked by Jak2V617F, a hallmark feature of PV. Re-expression of Stat5 in Stat5-deficient Jak2V617F knockin mice completely rescued the defects in transformation of hematopoietic progenitors and the PV phenotype. Together, these results indicate a critical function for Stat5 in the pathogenesis of PV. These findings also provide strong support for the development of Stat5 inhibitors as targeted therapies for the treatment of PV and other JAK2V617F-positive MPNs.
Figures
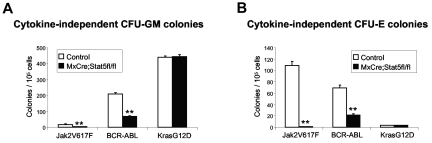
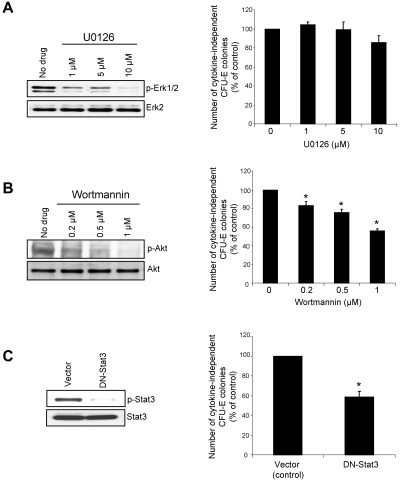
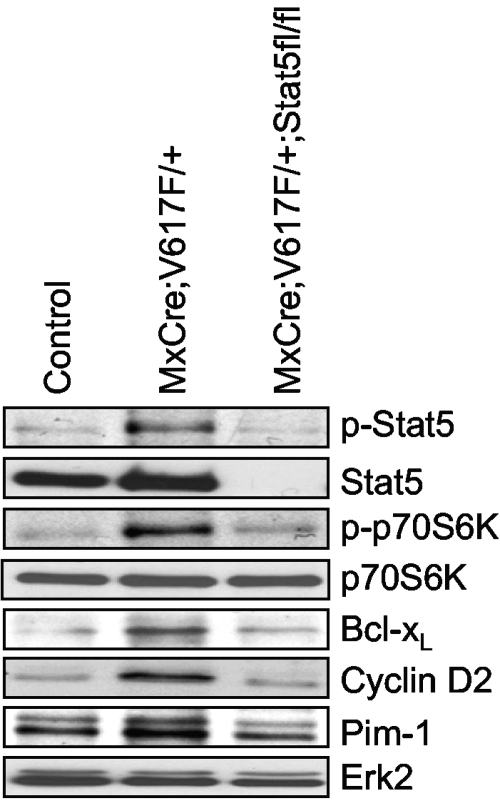
Comment in
-
Advancing the STATus of MPN pathogenesis.Blood. 2012 Apr 12;119(15):3374-6. doi: 10.1182/blood-2012-02-406611. Blood. 2012. PMID: 22500049
References
-
- James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signaling causes polycythemia vera. Nature. 2005;434(7037):1144–1148. - PubMed
-
- Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. - PubMed
-
- Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. - PubMed
-
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
