

MicroRNA-200c represses migration and invasion of breast cancer cells by targeting actin-regulatory proteins FHOD1 and PPM1F
- PMID: 22144583
- PMCID: PMC3266604
- DOI: 10.1128/MCB.06212-11
MicroRNA-200c represses migration and invasion of breast cancer cells by targeting actin-regulatory proteins FHOD1 and PPM1F
Abstract
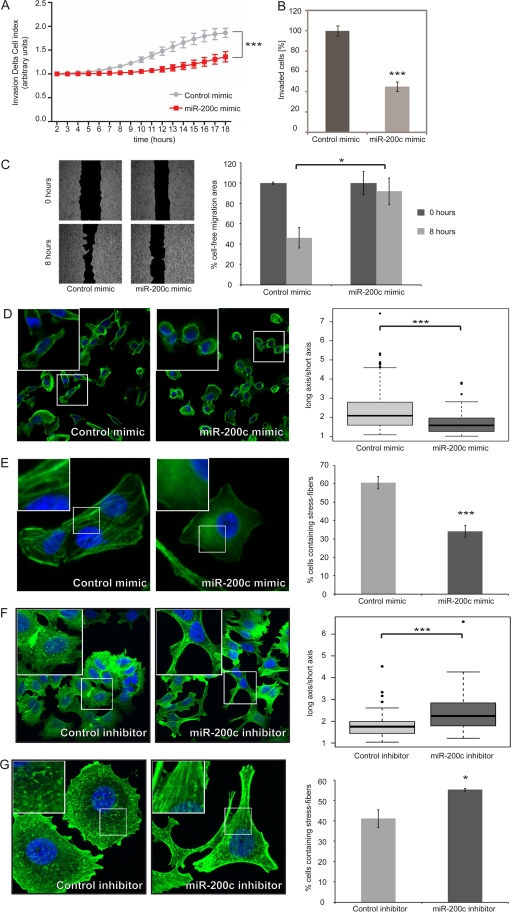
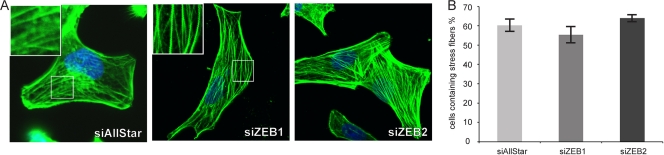
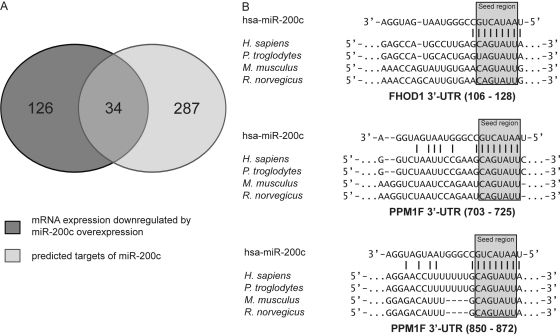
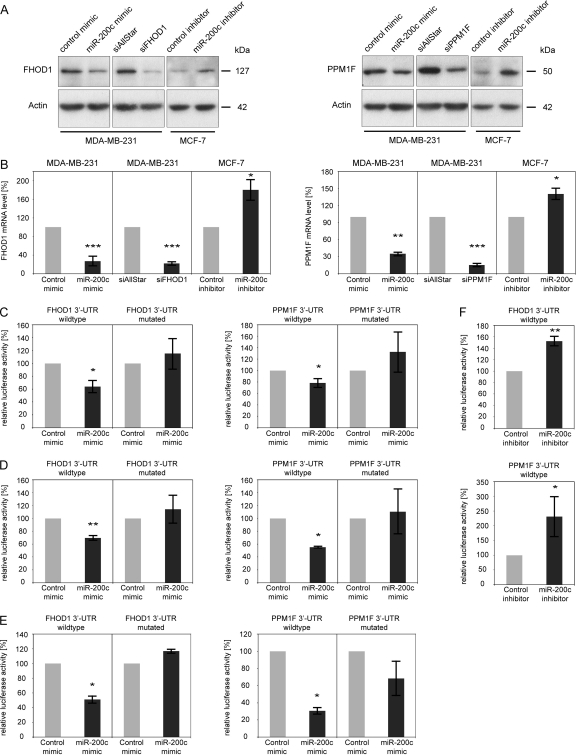
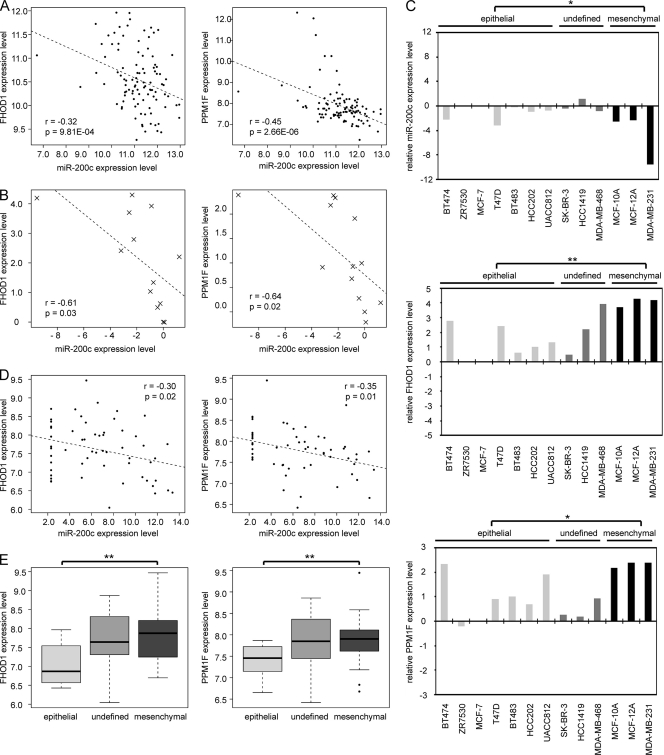
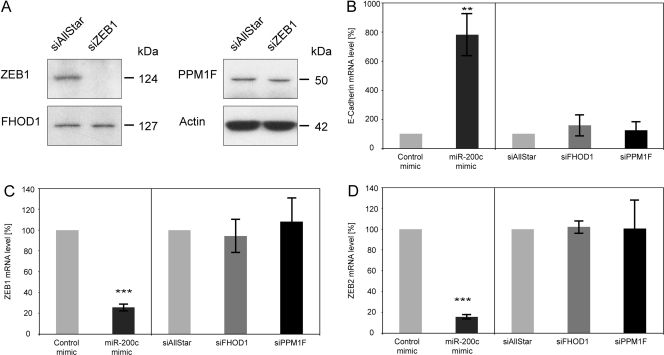
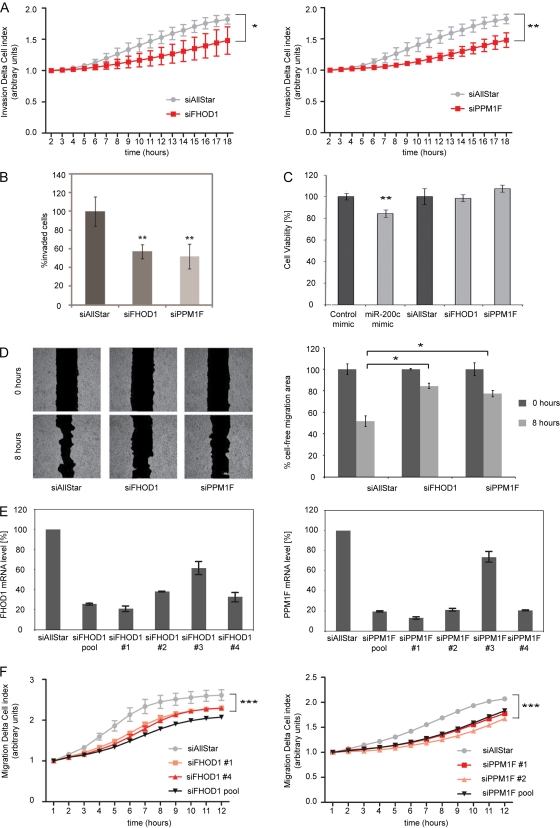
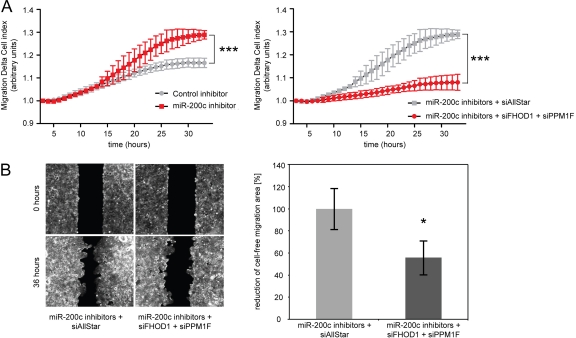
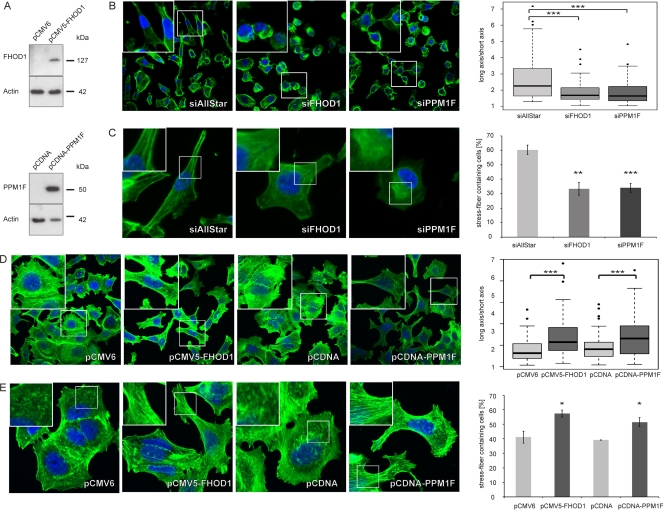
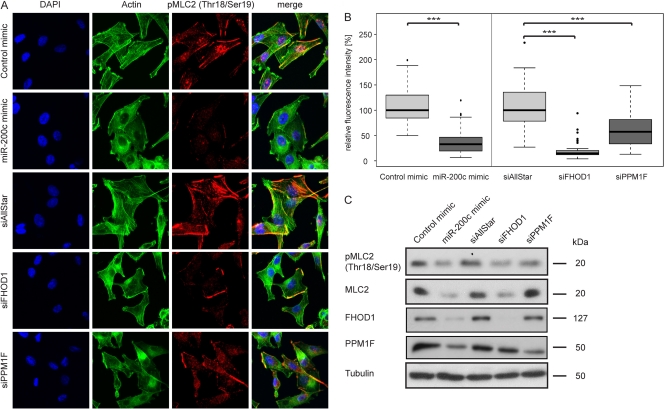
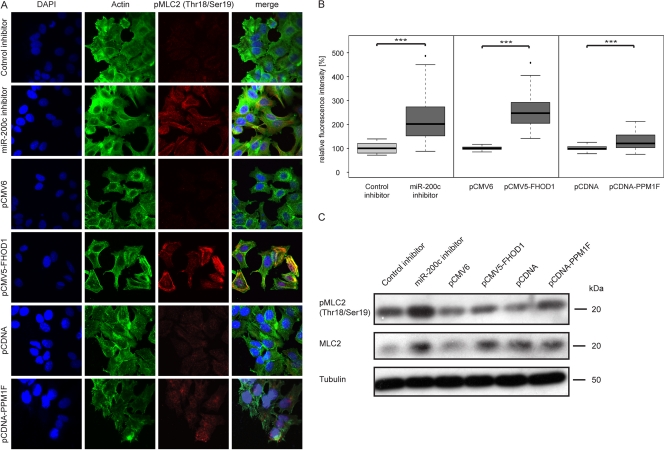
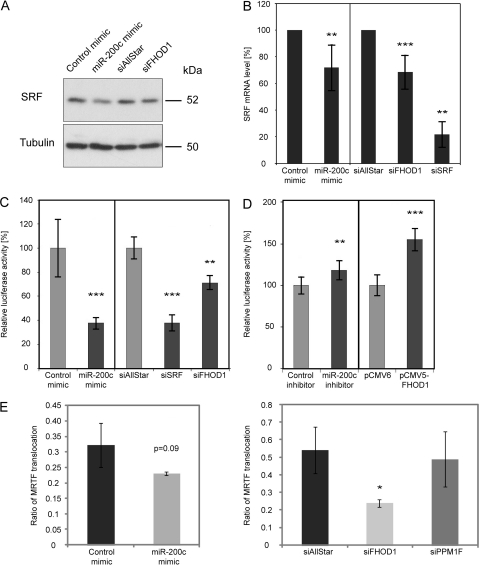
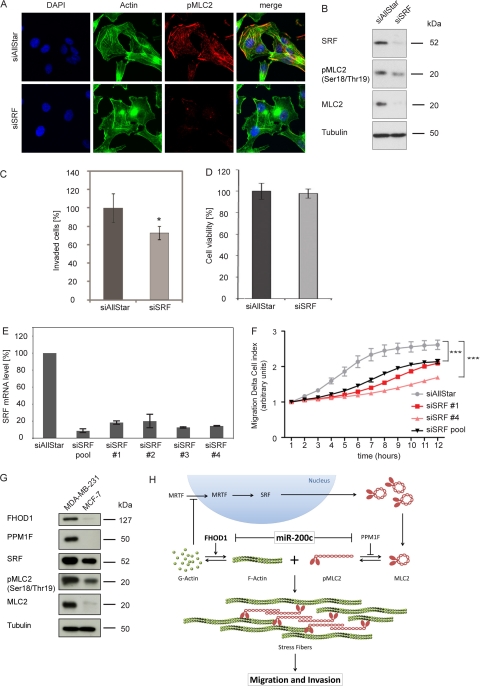
MicroRNA-200c (miR-200c) has been shown to suppress epithelial-mesenchymal transition (EMT), which is attributed mainly to targeting of ZEB1/ZEB2, repressors of the cell-cell contact protein E-cadherin. Here we demonstrated that modulation of miR-200c in breast cancer cells regulates cell migration, cell elongation, and transforming growth factor β (TGF-β)-induced stress fiber formation by impacting the reorganization of cytoskeleton that is independent of the ZEB/E-cadherin axis. We identified FHOD1 and PPM1F, direct regulators of the actin cytoskeleton, as novel targets of miR-200c. Remarkably, expression levels of FHOD1 and PPM1F were inversely correlated with the level of miR-200c in breast cancer cell lines, breast cancer patient samples, and 58 cancer cell lines of various origins. Furthermore, individual knockdown/overexpression of these target genes phenocopied the effects of miR-200c overexpression/inhibition on cell elongation, stress fiber formation, migration, and invasion. Mechanistically, targeting of FHOD1 by miR-200c resulted in decreased expression and transcriptional activity of serum response factor (SRF), mediated by interference with the translocation of the SRF coactivator mycocardin-related transcription factor A (MRTF-A). This finally led to downregulation of the expression and phosphorylation of the SRF target myosin light chain 2 (MLC2) gene, required for stress fiber formation and contractility. Thus, miR-200c impacts on metastasis by regulating several EMT-related processes, including a novel mechanism involving the direct targeting of actin-regulatory proteins.
Figures
References
-
- Blick T, et al. 2008. Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin. Exp. Metastasis 25:629–642 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous