

doi: 10.1371/journal.pcbi.1002277.
Epub 2011 Dec 1.
Computational mass spectrometry-based proteomics
Affiliations
- PMID: 22144880
- PMCID: PMC3228769
- DOI: 10.1371/journal.pcbi.1002277
Item in Clipboard
Computational mass spectrometry-based proteomics
PLoS Comput Biol.
2011 Dec.
No abstract available
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

The workflow requires a tight integration of biological and experimental (red) and computational and statistical (yellow) analysis steps.
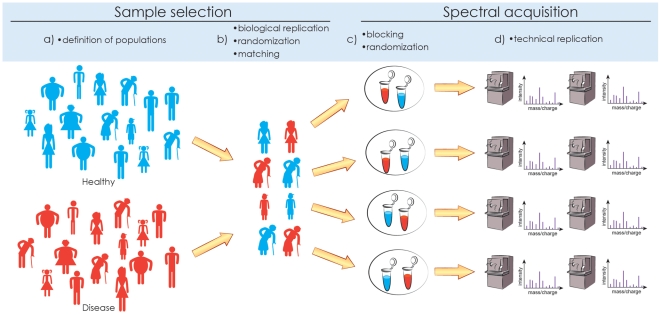
Statistical experimental design consists of (a) defining the populations of interest, (b) randomly selecting biological replicates from the population and (optionally) matching confounding factors, (c) randomly allocating biological samples to spectral acquisition and (optionally) grouping the samples in balanced blocks for joint profiling, and (d) (optionally) acquiring technical replicate measurements on the biological samples. Replication, randomization, and blocking are necessary to avoid biases and maximize the efficiency of the experiment.
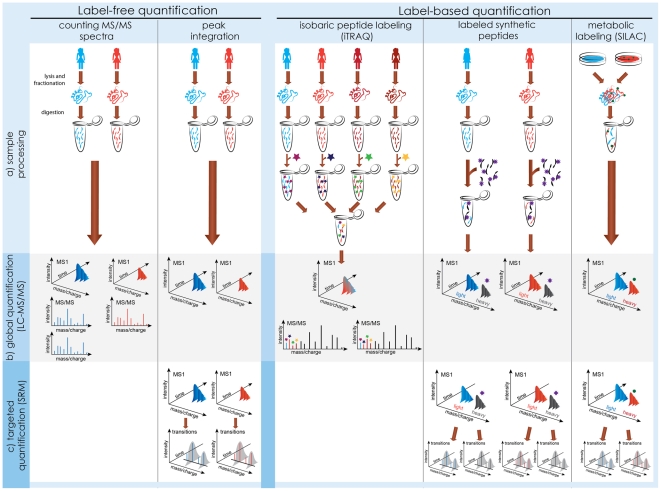
(a) Sample processing. Label-free quantification requires minimal sample manipulation, and acquires spectra from each sample in a separate mass spectrometry run. Label-based quantification varies in the timing and type of the labeling steps, but always simultaneously profiles two or more biological samples within a run. (b) Global label-free workflows achieve relative quantification by comparing counts of MS/MS spectra, or intensities of MS peaks between runs. Global label-based workflows compare intensities of reporter MS/MS fragments (iTRAQ) or MS peaks (SILAC, synthetic peptides). (c) Targeted workflows are an alternative to global quantification. They are most sensitive, but require an a priori knowledge of the proteins of interest, and of the technological characteristics of their peptides. Label-free targeted experiments compare intensities of transitions between runs, and label-based experiments within a run.
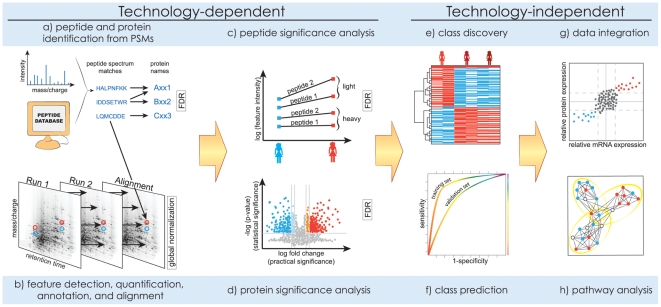
Analysis of the acquired spectra includes (a, b) signal processing, (c, d) significance analysis, and (e–h) downstream analysis. Methods in (a–d) must reflect the technological properties of the workflows. Methods in (e–h) are technology-independent and are similar to the analysis of gene expression microarrays, but their use is affected by uncertainty in protein identities and the incomplete sampling of the proteome.
References
-
- Beck M, Claassen M, Aebersold R. Comprehensive proteomics. Curr Opin Biotechnol. 2011;22:3–8. - PubMed
-
- de Godoy LMF, Olsen JV, Cox J, Nielsen ML, Hubner NC, et al. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature. 2008;455:1251. - PubMed
-
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. - PubMed
-
- Gavin AC, Bösche M, Krause R, Grandi P, Marzioch M, et al. Functional or ganization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. - PubMed
-
- Cox J, Mann M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu Rev Biochem. 2011;80:273–299. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
