

Charcot-Marie-Tooth-linked mutant GARS is toxic to peripheral neurons independent of wild-type GARS levels
- PMID: 22144914
- PMCID: PMC3228828
- DOI: 10.1371/journal.pgen.1002399
Charcot-Marie-Tooth-linked mutant GARS is toxic to peripheral neurons independent of wild-type GARS levels
Abstract
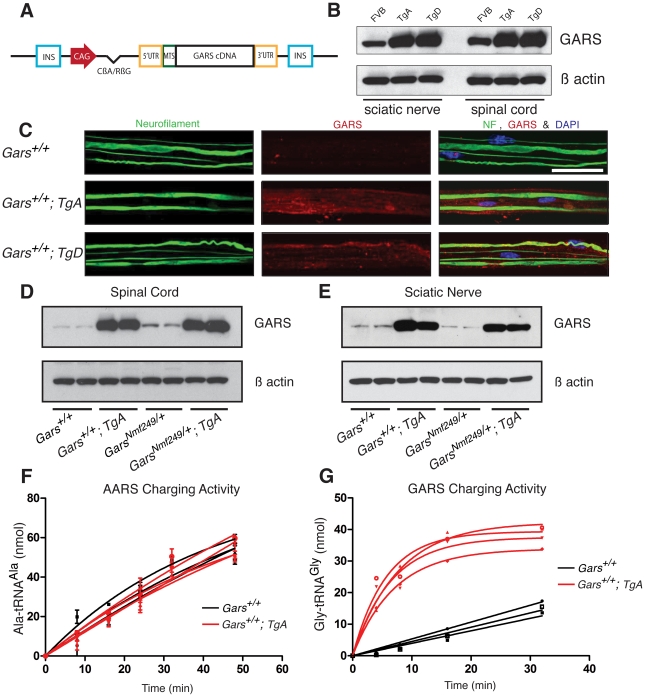
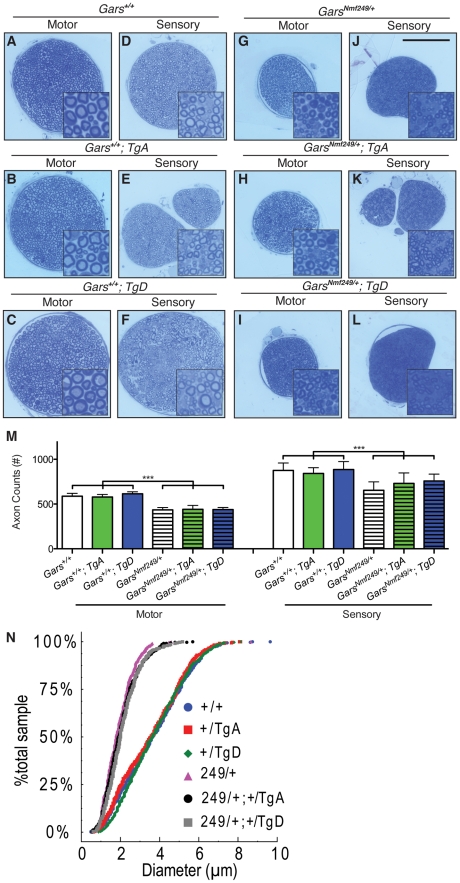
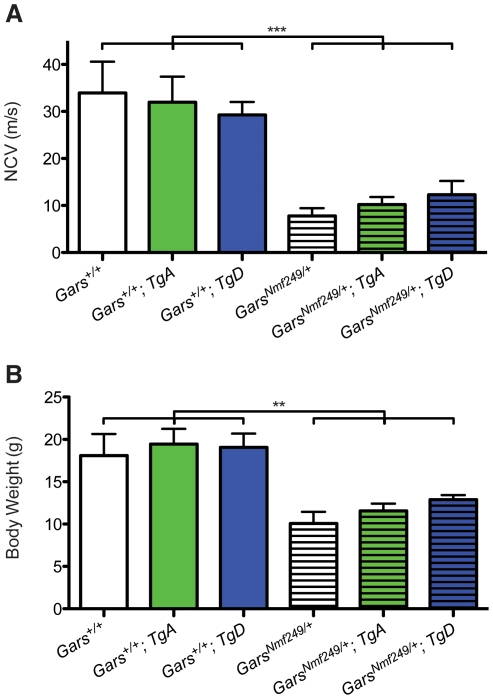
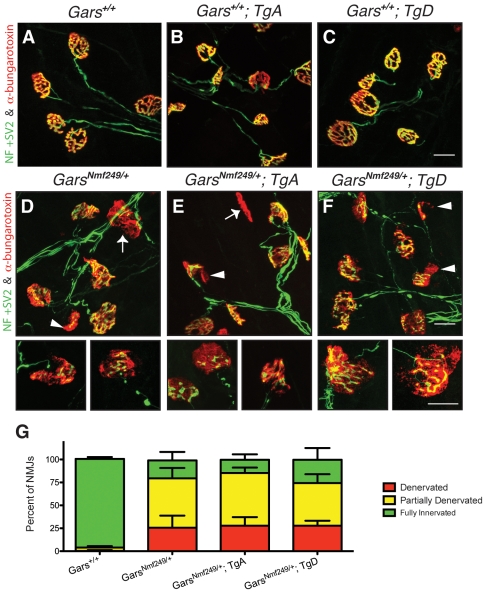
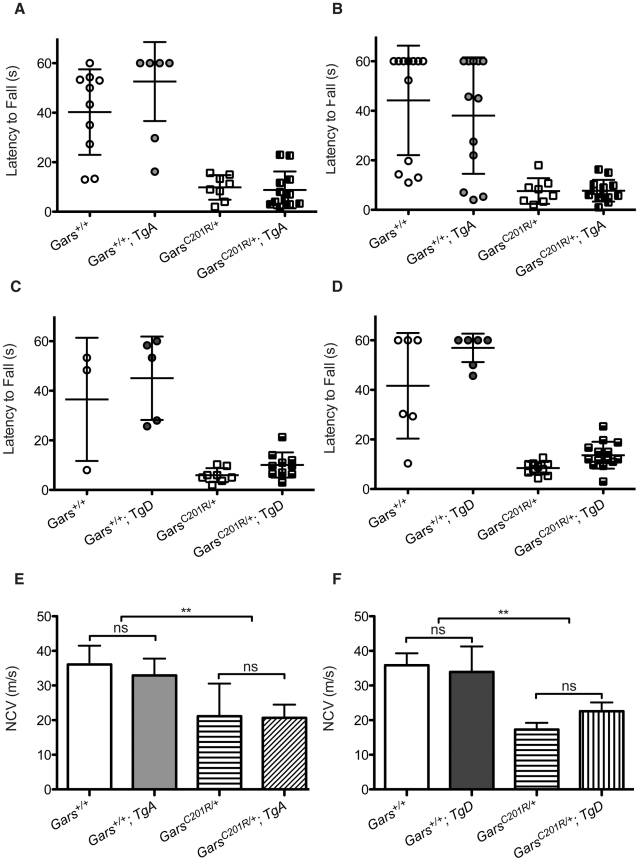
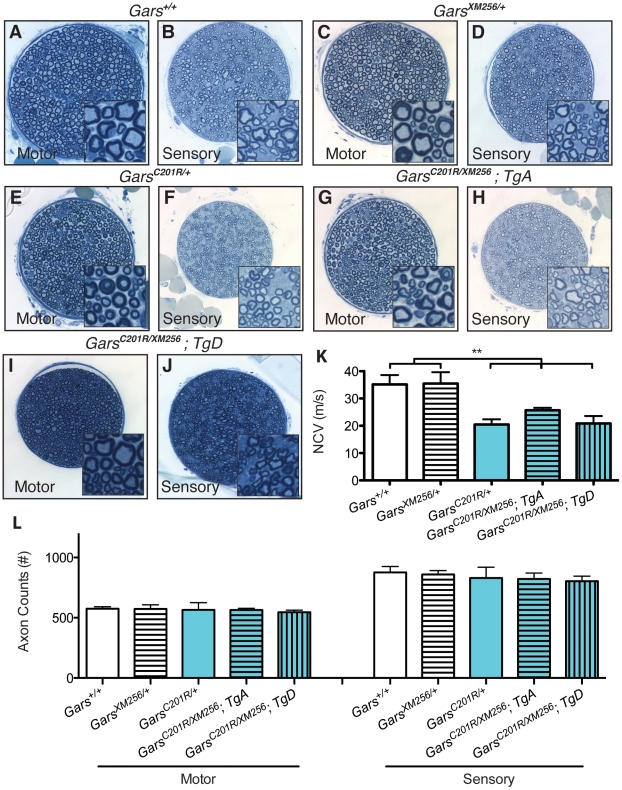
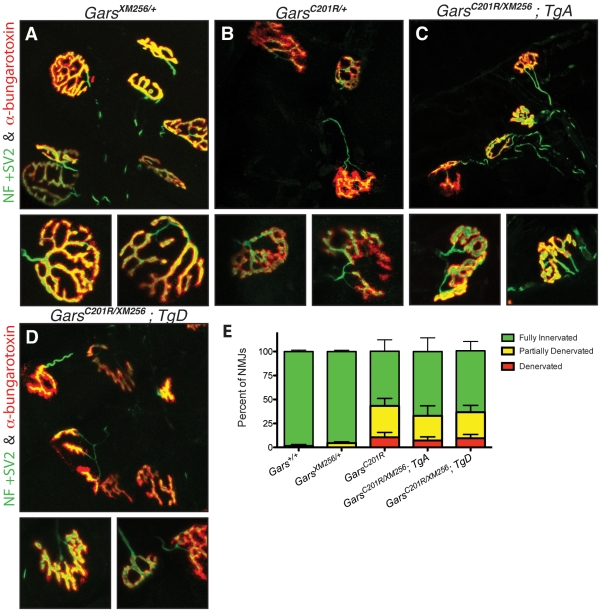
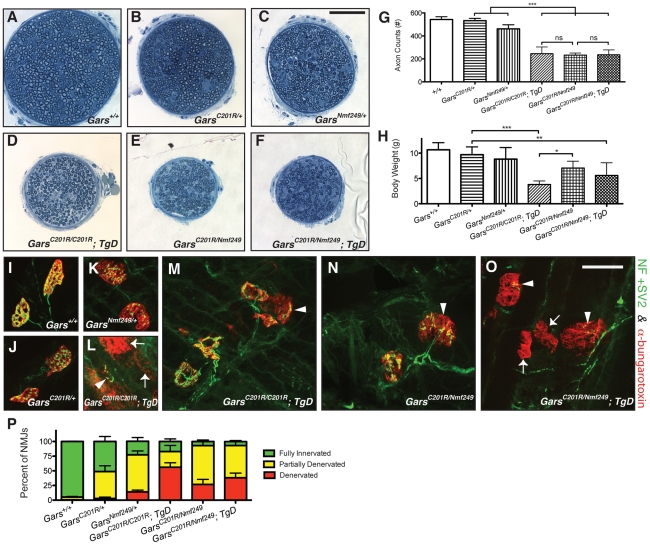
Charcot-Marie-Tooth disease type 2D (CMT2D) is a dominantly inherited peripheral neuropathy caused by missense mutations in the glycyl-tRNA synthetase gene (GARS). In addition to GARS, mutations in three other tRNA synthetase genes cause similar neuropathies, although the underlying mechanisms are not fully understood. To address this, we generated transgenic mice that ubiquitously over-express wild-type GARS and crossed them to two dominant mouse models of CMT2D to distinguish loss-of-function and gain-of-function mechanisms. Over-expression of wild-type GARS does not improve the neuropathy phenotype in heterozygous Gars mutant mice, as determined by histological, functional, and behavioral tests. Transgenic GARS is able to rescue a pathological point mutation as a homozygote or in complementation tests with a Gars null allele, demonstrating the functionality of the transgene and revealing a recessive loss-of-function component of the point mutation. Missense mutations as transgene-rescued homozygotes or compound heterozygotes have a more severe neuropathy than heterozygotes, indicating that increased dosage of the disease-causing alleles results in a more severe neurological phenotype, even in the presence of a wild-type transgene. We conclude that, although missense mutations of Gars may cause some loss of function, the dominant neuropathy phenotype observed in mice is caused by a dose-dependent gain of function that is not mitigated by over-expression of functional wild-type protein.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clinical Genetics 1974 - PubMed
-
- Jani-Acsadi A, Krajewski K, Shy ME. Charcot-Marie-Tooth neuropathies: diagnosis and management. Semin Neurol. 2008;28:185–194. - PubMed
-
- Nicholson G, Myers S. Intermediate forms of Charcot-Marie-Tooth neuropathy: a review. Neuromolecular Med. 2006;8:123–130. - PubMed
-
- Timmerman V, Nelis E, Van Hul W, Nieuwenhuijsen BW, Chen KL, et al. The peripheral myelin protein gene PMP-22 is contained within the Charcot-Marie-Tooth disease type 1A duplication. Nat Genet. 1992;1:171–175. - PubMed
-
- Hayasaka K, Himoro M, Sato W, Takada G, Uyemura K, et al. Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat Genet. 1993;5:31–34. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
