

Oncogenic EGFR signaling activates an mTORC2-NF-κB pathway that promotes chemotherapy resistance
- PMID: 22145100
- PMCID: PMC3229221
- DOI: 10.1158/2159-8290.CD-11-0124
Oncogenic EGFR signaling activates an mTORC2-NF-κB pathway that promotes chemotherapy resistance
Abstract
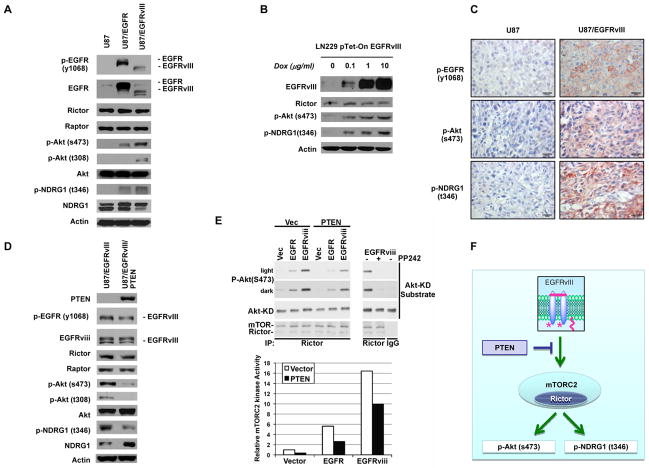
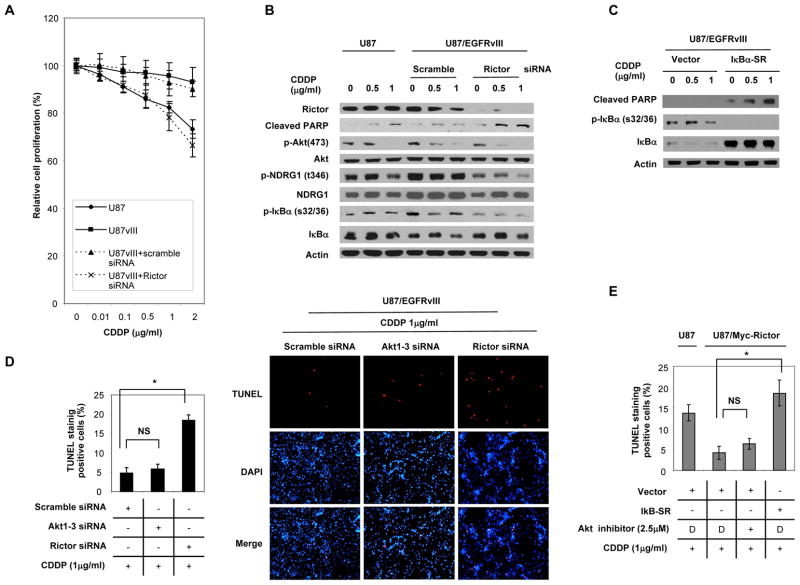
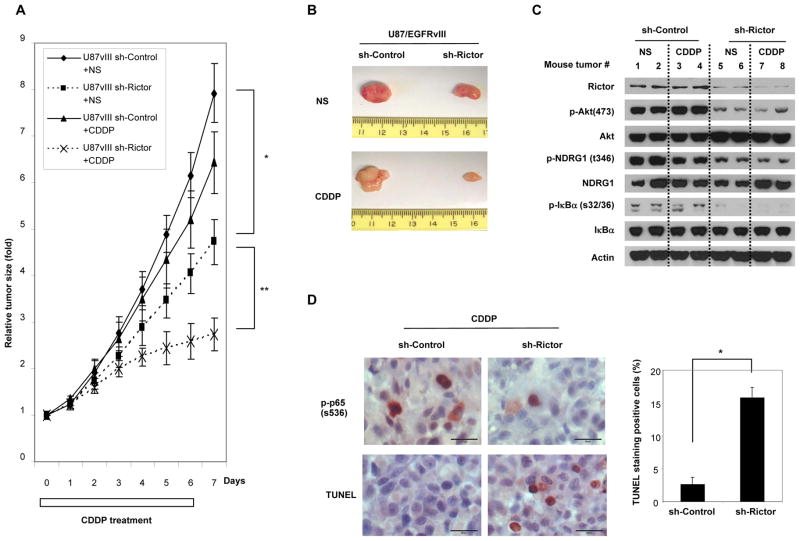
Although it is known that mTOR complex 2 (mTORC2) functions upstream of Akt, the role of this protein kinase complex in cancer is not well understood. Through an integrated analysis of cell lines, in vivo models, and clinical samples, we demonstrate that mTORC2 is frequently activated in glioblastoma (GBM), the most common malignant primary brain tumor of adults. We show that the common activating epidermal growth factor receptor (EGFR) mutation (EGFRvIII) stimulates mTORC2 kinase activity, which is partially suppressed by PTEN. mTORC2 signaling promotes GBM growth and survival and activates NF-κB. Importantly, this mTORC2-NF-κB pathway renders GBM cells and tumors resistant to chemotherapy in a manner independent of Akt. These results highlight the critical role of mTORC2 in the pathogenesis of GBM, including through the activation of NF-κB downstream of mutant EGFR, leading to a previously unrecognized function in cancer chemotherapy resistance. These findings suggest that therapeutic strategies targeting mTORC2, alone or in combination with chemotherapy, will be effective in the treatment of cancer.
Significance: This study demonstrates that EGFRvIII-activated mTORC2 signaling promotes GBM proliferation, survival, and chemotherapy resistance through Akt-independent activation of NF-κB. These results highlight the role of mTORC2 as an integrator of two canonical signaling networks that are commonly altered in cancer, EGFR/phosphoinositide-3 kinase (PI3K) and NF-κB. These results also validate the importance of mTORC2 as a cancer target and provide new insights into its role in mediating chemotherapy resistance, suggesting new treatment strategies.
Keywords: EGFRvIII; NF-κB; Rictor; and chomotherapy resistance; mTORC2.
© 2011 AACR.
Figures




Comment in
-
mTORC 2:1 for chemotherapy sensitization in glioblastoma.Cancer Discov. 2011 Nov;1(6):475-6. doi: 10.1158/2159-8290.CD-11-0264. Cancer Discov. 2011. PMID: 22586651
References
-
- Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2:pe24. - PubMed
-
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–18. - PubMed
-
- Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–71. - PubMed
-
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
