

Carbon monoxide inhibits Fas activating antibody-induced apoptosis in endothelial cells
- PMID: 22146483
- PMCID: PMC3231877
- DOI: 10.1186/2045-9912-1-8
Carbon monoxide inhibits Fas activating antibody-induced apoptosis in endothelial cells
Retraction in
-
Retraction: Carbon monoxide inhibits Fas activating antibody-induced apoptosis in endothelial cells.Med Gas Res. 2023 Oct-Dec;13(4):180. doi: 10.4103/2045-9912.374045. Med Gas Res. 2023. PMID: 37077123 Free PMC article.
Abstract
Background: The extrinsic apoptotic pathway initiates when a death ligand, such as the Fas ligand, interacts with its cell surface receptor (ie., Fas/CD95), forming a death-inducing signaling complex (DISC). The Fas-dependent apoptotic pathway has been implicated in several models of lung or vascular injury. Carbon monoxide, an enzymatic product of heme oxygenase-1, exerts antiapoptotic effects at low concentration in vitro and in vivo.
Methods: Using mouse lung endothelial cells (MLEC), we examined the antiapoptotic potential of carbon monoxide against apoptosis induced by the Fas/CD95-activating antibody (Jo2). Carbon monoxide was applied to cell cultures in vitro. The expression and/or activation of apoptosis-related proteins and signaling intermediates were determined using Western Immunoblot and co-immunoprecipitation assays. Cell death was monitored by lactate dehydrogenase (LDH) release assays. Statistical significance was determined by student T-test and a value of P < 0.05 was considered significant.
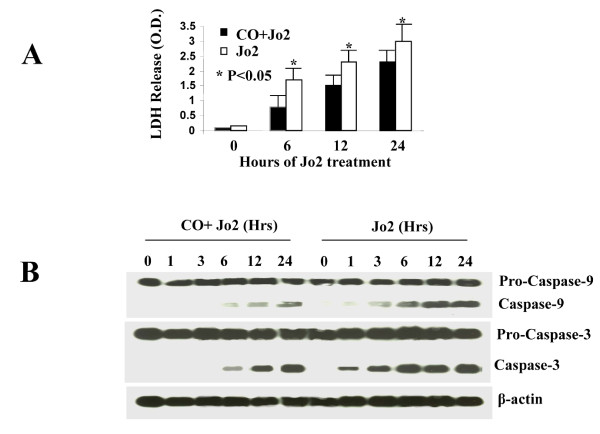
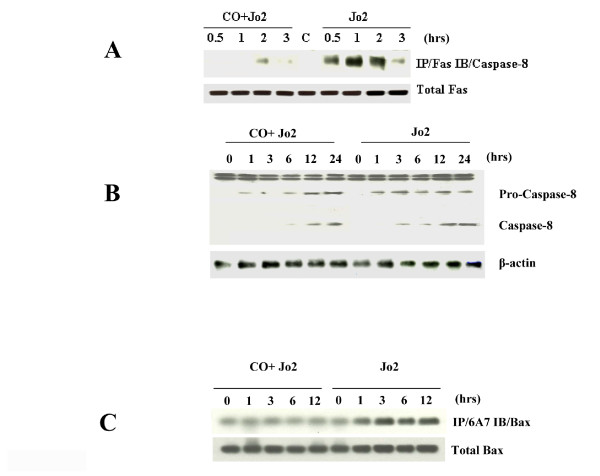
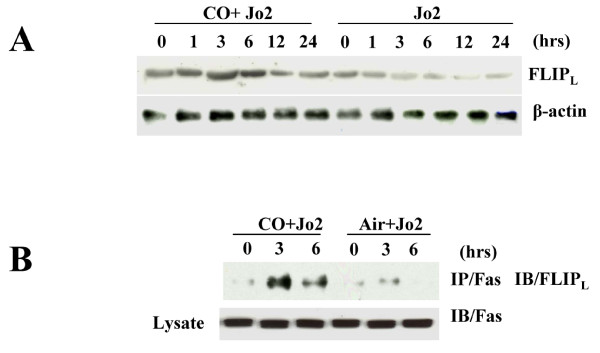
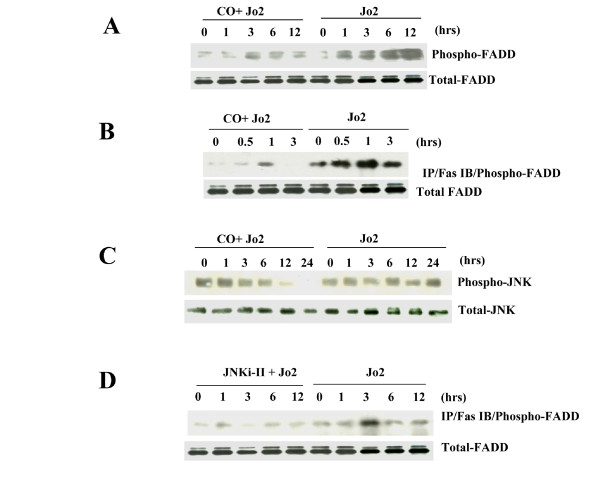
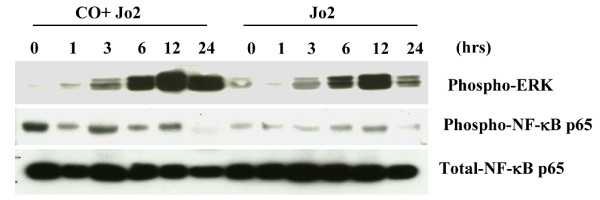
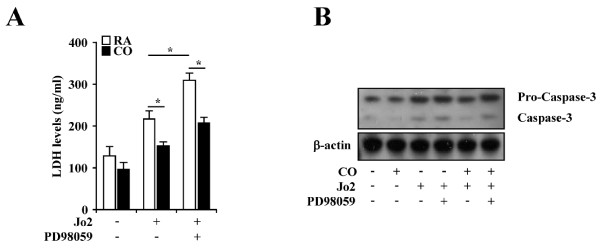
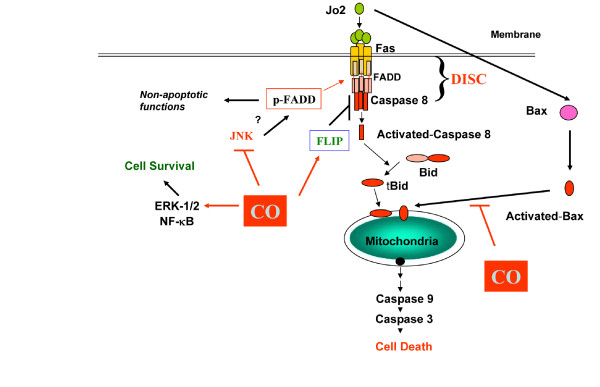
Results: Treatment of MLEC with Fas-activating antibody (Jo2) induced cell death associated with the formation of the DISC, and activation of caspases (-8, -9, and -3), as well as the pro-apoptotic Bcl-2 family protein Bax. Exposure of MLEC to carbon monoxide inhibited Jo2-induced cell death, which correlated with the inhibition of DISC formation, cleavage of caspases-8, -9, and -3, and Bax activation. Carbon monoxide inhibited the phosphorylation of the Fas-associated death domain-containing protein, as well as its association with the DISC. Furthermore, carbon monoxide induced the expression of the antiapoptotic protein FLIP and increased its association with the DISC.CO-dependent cytoprotection against Fas mediated apoptosis in MLEC depended in part on activation of ERK1/2-dependent signaling.
Conclusions: Carbon monoxide has been proposed as a potential therapy for lung and other diseases based in part on its antiapoptotic effects in endothelial cells. In vitro, carbon monoxide may inhibit both Fas/caspase-8 and Bax-dependent apoptotic signaling pathways induced by Fas-activating antibody in endothelial cells. Strategies to block Fas-dependent apoptotic pathways may be useful in development of therapies for lung or vascular disorders.
Figures
Similar articles
-
Carbon monoxide protects against hyperoxia-induced endothelial cell apoptosis by inhibiting reactive oxygen species formation.J Biol Chem. 2007 Jan 19;282(3):1718-26. doi: 10.1074/jbc.M607610200. Epub 2006 Nov 29. J Biol Chem. 2007. Retraction in: J Biol Chem. 2024 Mar;300(3):105758. doi: 10.1016/j.jbc.2024.105758. PMID: 17135272 Retracted.
-
Carbon monoxide promotes Fas/CD95-induced apoptosis in Jurkat cells.J Biol Chem. 2004 Oct 22;279(43):44327-34. doi: 10.1074/jbc.M406105200. Epub 2004 Jul 27. J Biol Chem. 2004. PMID: 15280387
-
Bcl-XL disrupts death-inducing signal complex formation in plasma membrane induced by hypoxia/reoxygenation.FASEB J. 2004 Dec;18(15):1826-33. doi: 10.1096/fj.04-2047com. FASEB J. 2004. PMID: 15576486
-
Regulation of CD95/Fas signaling at the DISC.Cell Death Differ. 2012 Jan;19(1):36-41. doi: 10.1038/cdd.2011.155. Epub 2011 Nov 11. Cell Death Differ. 2012. PMID: 22075988 Free PMC article. Review.
-
Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra.J Neuropathol Exp Neurol. 2003 Apr;62(4):329-39. doi: 10.1093/jnen/62.4.329. J Neuropathol Exp Neurol. 2003. PMID: 12722825 Review.
Cited by
-
D4F alleviates macrophage-derived foam cell apoptosis by inhibiting the NF-κB-dependent Fas/FasL pathway.Sci Rep. 2017 Aug 4;7(1):7333. doi: 10.1038/s41598-017-07656-0. Sci Rep. 2017. PMID: 28779128 Free PMC article.
-
Anti-apoptotic properties of carbon monoxide in porcine oocyte during in vitro aging.PeerJ. 2017 Oct 6;5:e3876. doi: 10.7717/peerj.3876. eCollection 2017. PeerJ. 2017. PMID: 29018614 Free PMC article.
-
Carbon monoxide in lung cell physiology and disease.Am J Physiol Cell Physiol. 2018 Feb 1;314(2):C211-C227. doi: 10.1152/ajpcell.00022.2017. Epub 2017 Nov 8. Am J Physiol Cell Physiol. 2018. PMID: 29118026 Free PMC article. Review.
-
Upregulation of heat shock protein 32 in Sertoli cells alleviates the impairments caused by heat shock-induced apoptosis in mouse testis.Cell Stress Chaperones. 2013 May;18(3):333-51. doi: 10.1007/s12192-012-0385-8. Epub 2012 Nov 28. Cell Stress Chaperones. 2013. PMID: 23188493 Free PMC article.
-
Carbon monoxide: present and future indications for a medical gas.Korean J Intern Med. 2013 Mar;28(2):123-40. doi: 10.3904/kjim.2013.28.2.123. Epub 2013 Feb 27. Korean J Intern Med. 2013. PMID: 23525151 Free PMC article. Review.
References
-
- Matute-Bello G, Liles WC, Steinberg KP, Kiener PA, Mongovin S, Chi EY, Jonas M, Martin TR. Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS) J Immunol. 1999;163:2217–2225. - PubMed
-
- Imai Y, Parodo J, Kajikawa O, de Perrot M, Fischer S, Edwards V, Cutz E, Liu M, Keshavjee S, Martin TR, Marshall JC, Ranieri VM, Slutsky AS. Injurious mechanical ventilation and end-organ epithelial cell apoptosis and organ dysfunction in an experimental model of acute respiratory distress syndrome. JAMA. 2003;289:2104–2112. doi: 10.1001/jama.289.16.2104. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
