

GAGE: A critical evaluation of genome assemblies and assembly algorithms
- PMID: 22147368
- PMCID: PMC3290791
- DOI: 10.1101/gr.131383.111
GAGE: A critical evaluation of genome assemblies and assembly algorithms
Erratum in
- Genome Res. 2012 Jun;22(6):1196
Abstract
New sequencing technology has dramatically altered the landscape of whole-genome sequencing, allowing scientists to initiate numerous projects to decode the genomes of previously unsequenced organisms. The lowest-cost technology can generate deep coverage of most species, including mammals, in just a few days. The sequence data generated by one of these projects consist of millions or billions of short DNA sequences (reads) that range from 50 to 150 nt in length. These sequences must then be assembled de novo before most genome analyses can begin. Unfortunately, genome assembly remains a very difficult problem, made more difficult by shorter reads and unreliable long-range linking information. In this study, we evaluated several of the leading de novo assembly algorithms on four different short-read data sets, all generated by Illumina sequencers. Our results describe the relative performance of the different assemblers as well as other significant differences in assembly difficulty that appear to be inherent in the genomes themselves. Three overarching conclusions are apparent: first, that data quality, rather than the assembler itself, has a dramatic effect on the quality of an assembled genome; second, that the degree of contiguity of an assembly varies enormously among different assemblers and different genomes; and third, that the correctness of an assembly also varies widely and is not well correlated with statistics on contiguity. To enable others to replicate our results, all of our data and methods are freely available, as are all assemblers used in this study.
Figures
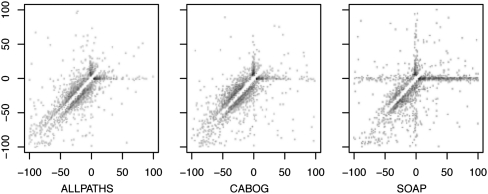
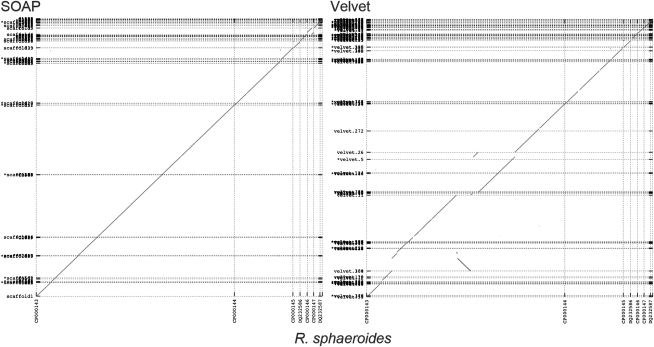
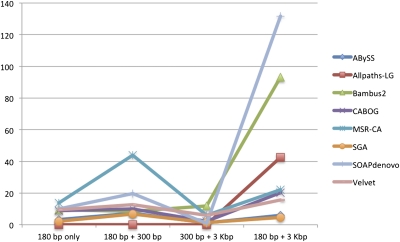
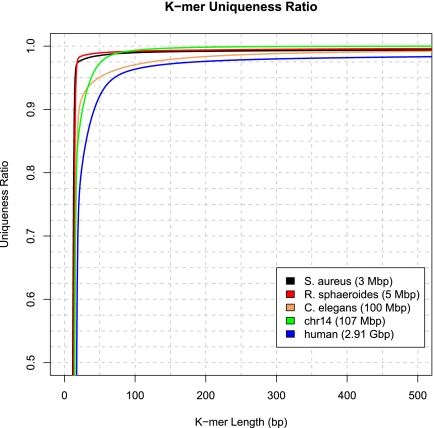
References
-
- Dalloul RA, Long JA, Zimin AV, Aslam L, Beal K, Blomberg Le A, Bouffard P, Burt DW, Crasta O, Crooijmans RP, et al. 2010. Multi-platform next-generation sequencing of the domestic turkey (Meleagris gallopavo): Genome assembly and analysis. PLoS Biol 8: e1000475 doi: 10.1371/journal.pbio.1000475 - PMC - PubMed
-
- Ju YS, Kim JI, Kim S, Hong D, Park H, Shin JY, Lee S, Lee WC, Yu SB, Park SS, et al. 2011. Extensive genomic and transcriptional diversity identified through massively parallel DNA and RNA sequencing of eighteen Korean individuals. Nat Genet 43: 745–752 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials