

Autophagy in lysosomal myopathies
- PMID: 22150923
- PMCID: PMC8029060
- DOI: 10.1111/j.1750-3639.2011.00543.x
Autophagy in lysosomal myopathies
Abstract
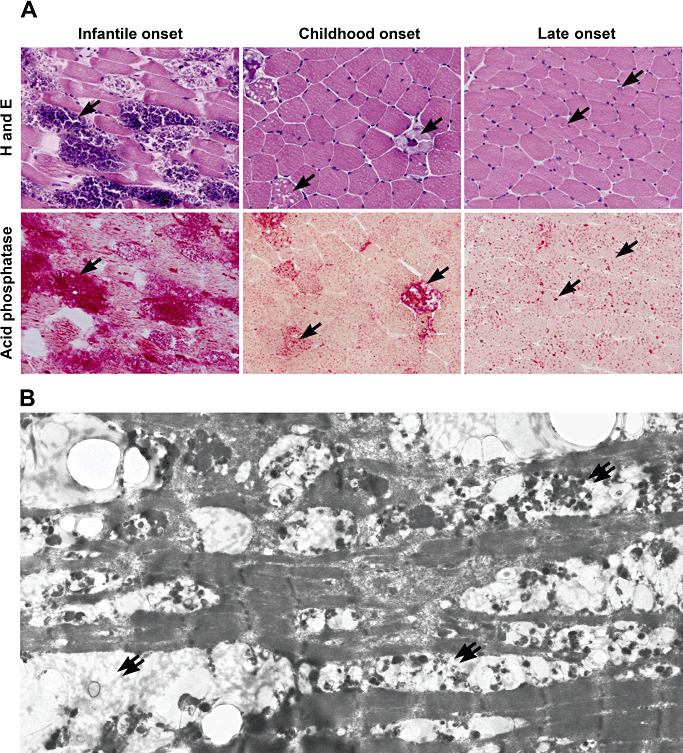
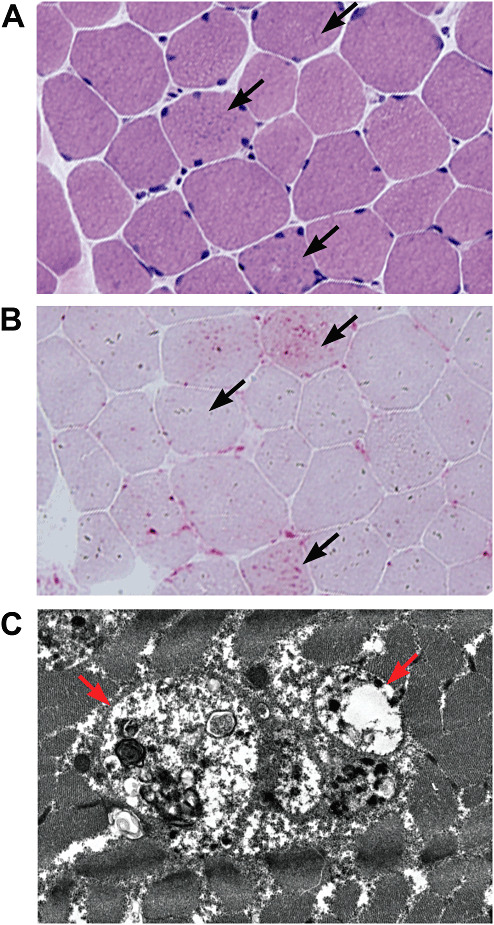
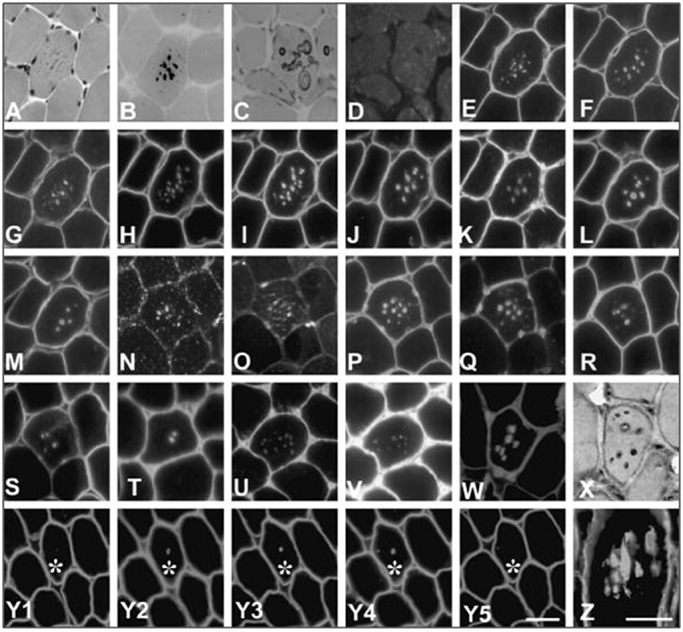
Lysosomal myopathies are hereditary myopathies characterized morphologically by the presence of autophagic vacuoles. In mammals, autophagy plays an important role for the turnover of cellular components, particularly in response to starvation or glucagons. In normal muscle, autolysosomes or autophagosomes are typically inconspicuous. In distinct neuromuscular disorders, however, lysosomes become structurally abnormal and functionally impaired, leading to the accumulation of autophagic vacuoles in myofibers. In some instances, the accumulation of autophagic vacuoles can be a prominent feature, implicating autophagy as a contributor to disease pathomechanism and/or progression. At present, there are two disorders in the muscle that are associated with a primary defect in lysosomal proteins, namely Pompe disease and Danon disease. This review will give a brief discussion on these disorders, highlighting the role of autophagy in disease progression.
© 2011 The Authors; Brain Pathology © 2011 International Society of Neuropathology.
Figures
References
-
- Akanas V, Engel WK (2006) Inclusion‐body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition. Neurology 66:S39–S48. - PubMed
-
- Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL et al (2001) Recombinant human acid alpha‐glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med 3:132–138. - PubMed
-
- Ballabio A, Gieselmann V (2009) Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta 1793:684–696. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
