

Consistency-based detection of potential tumor-specific deletions in matched normal/tumor genomes
- PMID: 22152084
- PMCID: PMC3283309
- DOI: 10.1186/1471-2105-12-S9-S21
Consistency-based detection of potential tumor-specific deletions in matched normal/tumor genomes
Abstract
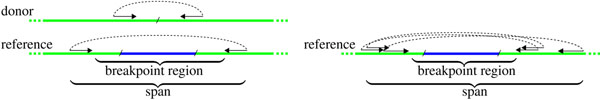
Background: Structural variations in human genomes, such as insertions, deletion, or rearrangements, play an important role in cancer development. Next-Generation Sequencing technologies have been central in providing ways to detect such variations. Most existing methods however are limited to the analysis of a single genome, and it is only recently that the comparison of closely related genomes has been considered. In particular, a few recent works considered the analysis of data sets obtained by sequencing both tumor and healthy tissues of the same cancer patient. In that context, the goal is to detect variations that are specific to exactly one of the genomes, for example to differentiate between patient-specific and tumor-specific variations. This is a difficult task, especially when facing the additional challenge of the possible contamination of healthy tissues by tumor cells and conversely.
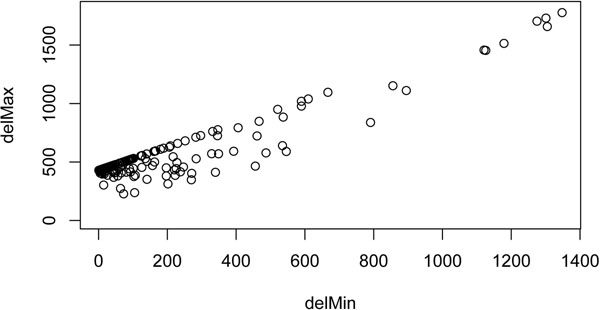
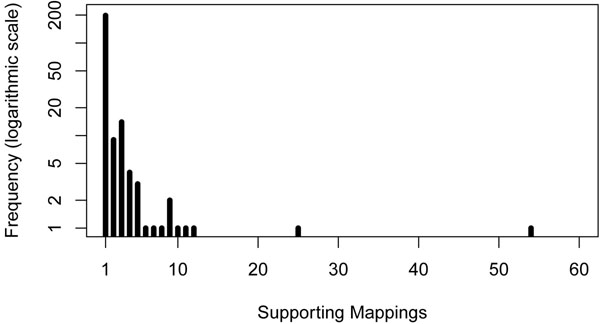
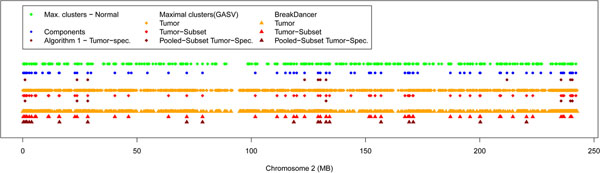
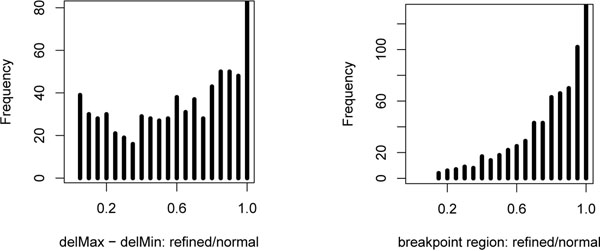
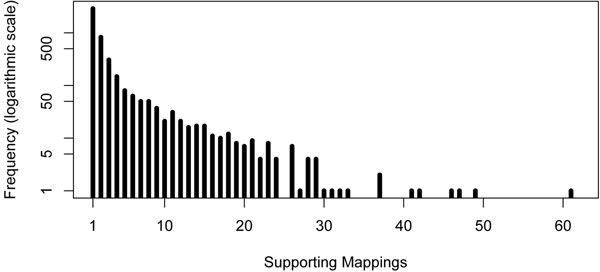
Results: In the current work, we analyzed a data set of paired-end short-reads, obtained by sequencing tumor tissues and healthy tissues, both from the same cancer patient. Based on a combinatorial notion of conflict between deletions, we show that in the tumor data, more deletions are predicted than there could actually be in a diploid genome. In contrast, the predictions for the data from normal tissues are almost conflict-free. We designed and applied a method, specific to the analysis of such pooled and contaminated data sets, to detect potential tumor-specific deletions. Our method takes the deletion calls from both data sets and assigns reads from the mixed tumor/normal data to the normal one with the goal to minimize the number of reads that need to be discarded to obtain a set of conflict-free deletion clusters. We observed that, on the specific data set we analyze, only a very small fraction of the reads needs to be discarded to obtain a set of consistent deletions.
Conclusions: We present a framework based on a rigorous definition of consistency between deletions and the assumption that the tumor sample also contains normal cells. A combined analysis of both data sets based on this model allowed a consistent explanation of almost all data, providing a detailed picture of candidate patient- and tumor-specific deletions.
Figures


Similar articles
-
Unraveling overlapping deletions by agglomerative clustering.BMC Genomics. 2013;14 Suppl 1(Suppl 1):S12. doi: 10.1186/1471-2164-14-S1-S12. Epub 2013 Jan 21. BMC Genomics. 2013. PMID: 23369161 Free PMC article.
-
iSVP: an integrated structural variant calling pipeline from high-throughput sequencing data.BMC Syst Biol. 2013;7 Suppl 6(Suppl 6):S8. doi: 10.1186/1752-0509-7-S6-S8. Epub 2013 Dec 13. BMC Syst Biol. 2013. PMID: 24564972 Free PMC article.
-
Identification of genomic indels and structural variations using split reads.BMC Genomics. 2011 Jul 25;12:375. doi: 10.1186/1471-2164-12-375. BMC Genomics. 2011. PMID: 21787423 Free PMC article.
-
Analysis of next-generation genomic data in cancer: accomplishments and challenges.Hum Mol Genet. 2010 Oct 15;19(R2):R188-96. doi: 10.1093/hmg/ddq391. Epub 2010 Sep 15. Hum Mol Genet. 2010. PMID: 20843826 Free PMC article. Review.
-
Progress in the detection of human genome structural variations.Sci China C Life Sci. 2009 Jun;52(6):560-7. doi: 10.1007/s11427-009-0078-4. Epub 2009 Jun 26. Sci China C Life Sci. 2009. PMID: 19557334 Review.
Cited by
-
An integrative probabilistic model for identification of structural variation in sequencing data.Genome Biol. 2012;13(3):R22. doi: 10.1186/gb-2012-13-3-r22. Genome Biol. 2012. PMID: 22452995 Free PMC article.
-
Unraveling overlapping deletions by agglomerative clustering.BMC Genomics. 2013;14 Suppl 1(Suppl 1):S12. doi: 10.1186/1471-2164-14-S1-S12. Epub 2013 Jan 21. BMC Genomics. 2013. PMID: 23369161 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
