

Thiamine pyrophosphokinase deficiency in encephalopathic children with defects in the pyruvate oxidation pathway
- PMID: 22152682
- PMCID: PMC3234371
- DOI: 10.1016/j.ajhg.2011.11.007
Thiamine pyrophosphokinase deficiency in encephalopathic children with defects in the pyruvate oxidation pathway
Abstract
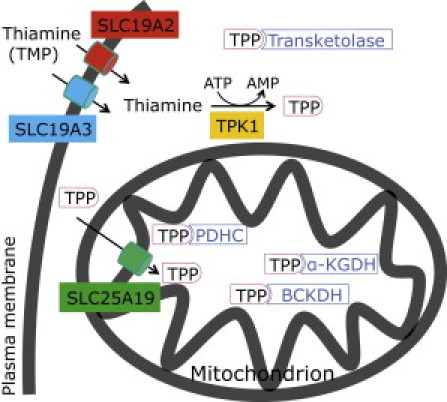
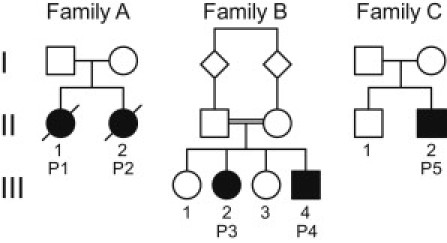
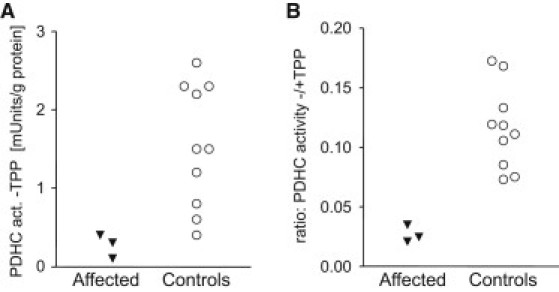
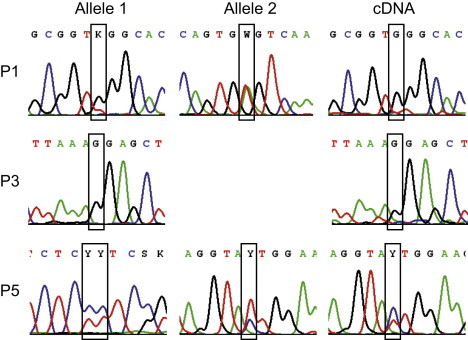
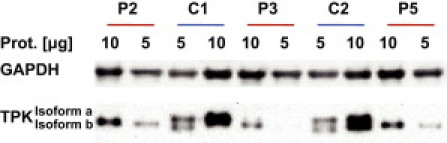
Thiamine pyrophosphate (TPP) is an essential cofactor of the cytosolic transketolase and of three mitochondrial enzymes involved in the oxidative decarboxylation of either pyruvate, α-ketoglutarate or branched chain amino acids. Thiamine is taken up by specific transporters into the cell and converted to the active TPP by thiamine pyrophosphokinase (TPK) in the cytosol from where it can be transported into mitochondria. Here, we report five individuals from three families presenting with variable degrees of ataxia, psychomotor retardation, progressive dystonia, and lactic acidosis. Investigation of the mitochondrial energy metabolism showed reduced oxidation of pyruvate but normal pyruvate dehydrogenase complex activity in the presence of excess TPP. A reduced concentration of TPP was found in the muscle and blood. Mutation analysis of TPK1 uncovered three missense, one splice-site, and one frameshift mutation resulting in decreased TPK protein levels.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Labay V., Raz T., Baron D., Mandel H., Williams H., Barrett T., Szargel R., McDonald L., Shalata A., Nosaka K., et al. Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat. Genet. 1999;22:300–304. - PubMed
-
- Ozand P.T., Gascon G.G., Al Essa M., Joshi S., Al Jishi E., Bakheet S., Al Watban J., Al-Kawi M.Z., Dabbagh O. Biotin-responsive basal ganglia disease: A novel entity. Brain. 1998;121:1267–1279. - PubMed
-
- Subramanian V.S., Marchant J.S., Said H.M. Biotin-responsive basal ganglia disease-linked mutations inhibit thiamine transport via hTHTR2: Biotin is not a substrate for hTHTR2. Am. J. Physiol. Cell Physiol. 2006;291:C851–C859. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
