

Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins
- PMID: 22153072
- PMCID: PMC3319069
- DOI: 10.1016/j.cell.2011.10.053
Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins
Abstract
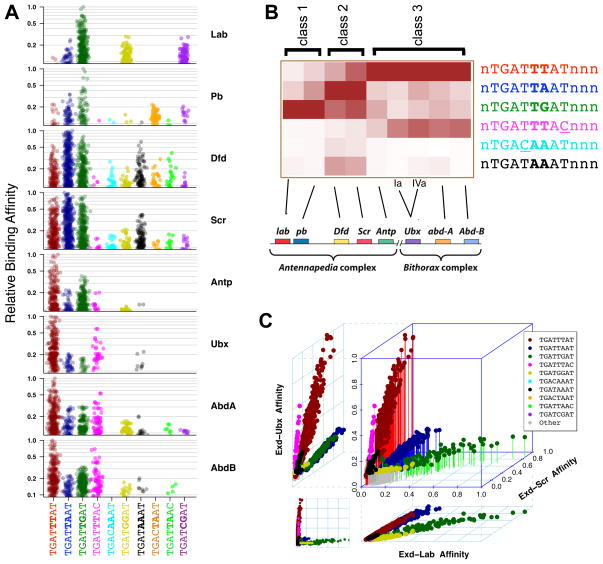
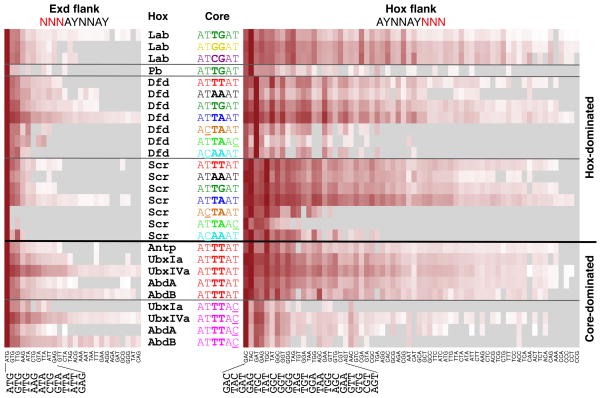
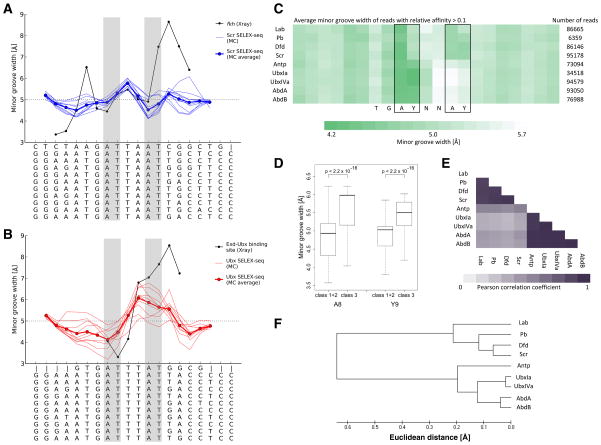
Members of transcription factor families typically have similar DNA binding specificities yet execute unique functions in vivo. Transcription factors often bind DNA as multiprotein complexes, raising the possibility that complex formation might modify their DNA binding specificities. To test this hypothesis, we developed an experimental and computational platform, SELEX-seq, that can be used to determine the relative affinities to any DNA sequence for any transcription factor complex. Applying this method to all eight Drosophila Hox proteins, we show that they obtain novel recognition properties when they bind DNA with the dimeric cofactor Extradenticle-Homothorax (Exd). Exd-Hox specificities group into three main classes that obey Hox gene collinearity rules and DNA structure predictions suggest that anterior and posterior Hox proteins prefer DNA sequences with distinct minor groove topographies. Together, these data suggest that emergent DNA recognition properties revealed by interactions with cofactors contribute to transcription factor specificities in vivo.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures




Comment in
-
A partner evokes latent differences between Hox proteins.Cell. 2011 Dec 9;147(6):1220-1. doi: 10.1016/j.cell.2011.11.046. Cell. 2011. PMID: 22153067
-
Gene regulation: Resolving transcription factor binding.Nat Rev Genet. 2011 Dec 29;13(2):71. doi: 10.1038/nrg3153. Nat Rev Genet. 2011. PMID: 22207166 No abstract available.
References
-
- Alexander T, Nolte C, Krumlauf R. Hox genes and segmentation of the hindbrain and axial skeleton. Annu Rev Cell Dev Biol. 2009;25:431–456. - PubMed
-
- Bartfeld D, Shimon L, Couture GC, Rabinovich D, Frolow F, Levanon D, Groner Y, Shakked Z. DNA recognition by the RUNX1 transcription factor is mediated by an allosteric transition in the RUNT domain and by DNA bending. Structure. 2002;10:1395–1407. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
