

Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution
- PMID: 22153082
- PMCID: PMC3243364
- DOI: 10.1016/j.cell.2011.11.013
Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution
Abstract
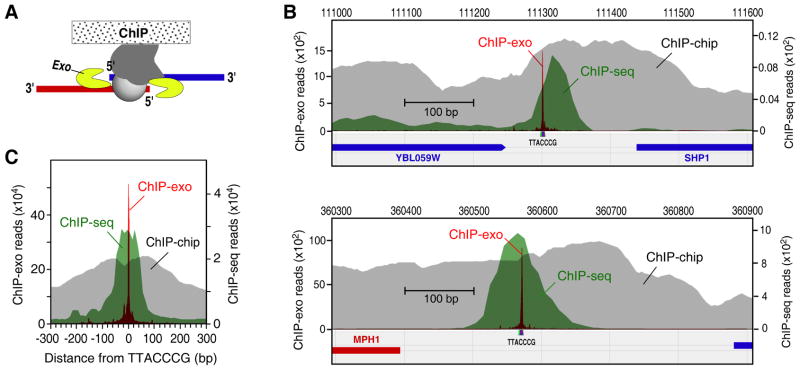
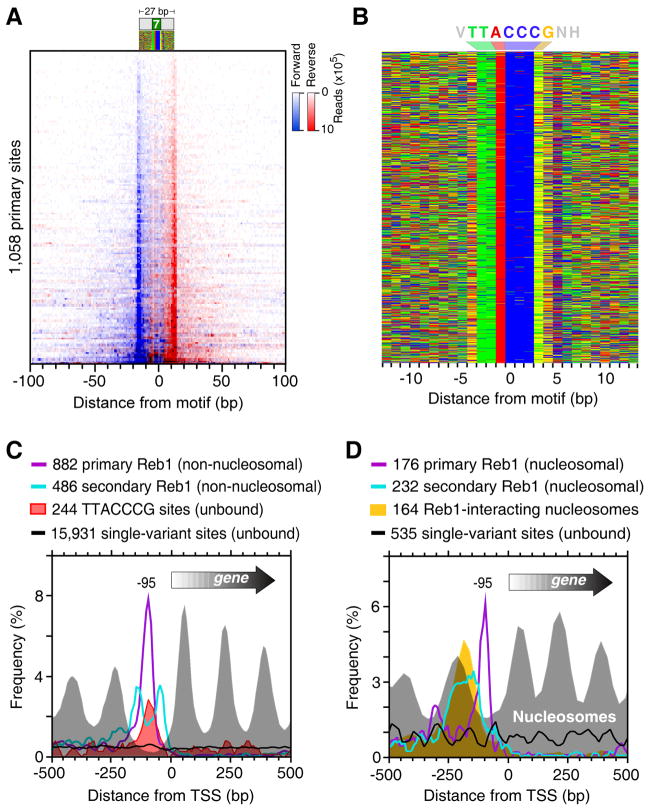
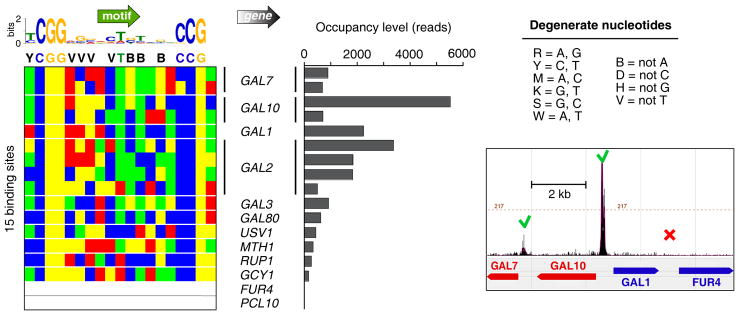
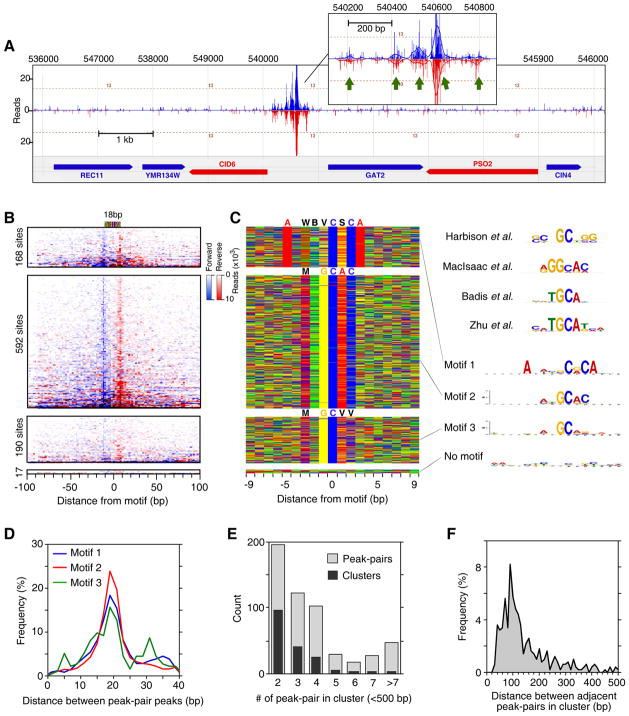
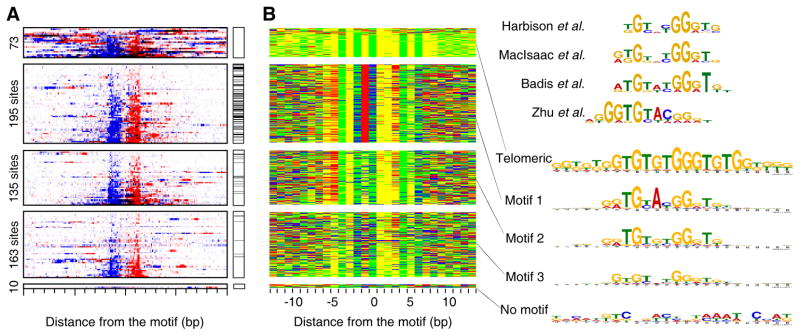
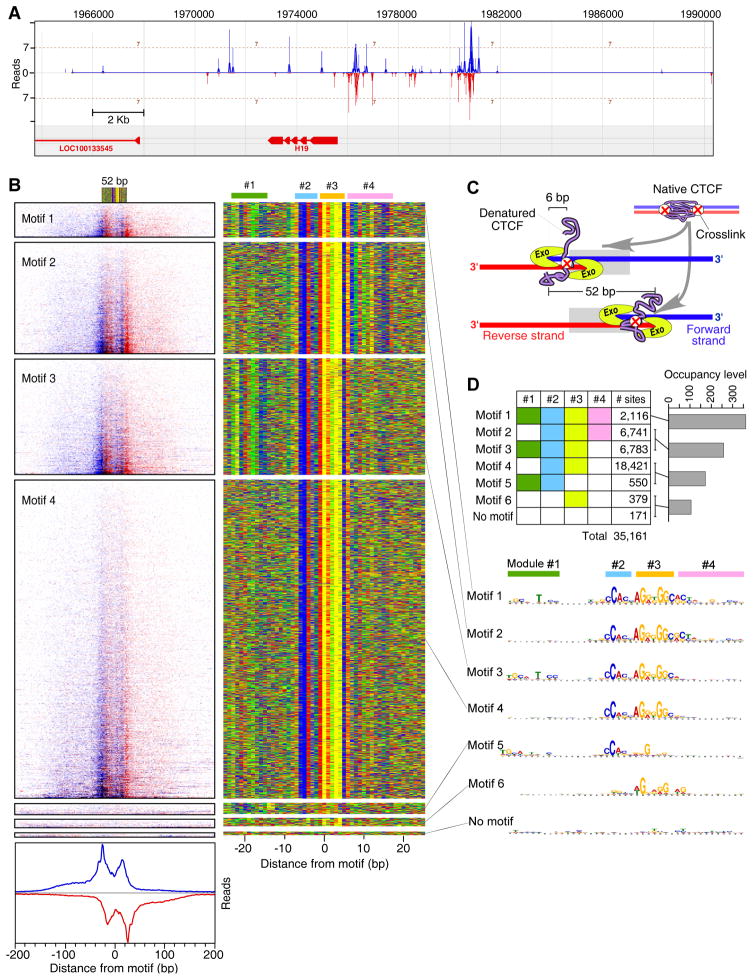
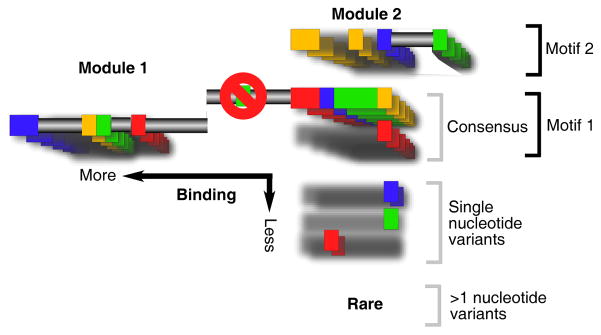
Chromatin immunoprecipitation (ChIP-chip and ChIP-seq) assays identify where proteins bind throughout a genome. However, DNA contamination and DNA fragmentation heterogeneity produce false positives (erroneous calls) and imprecision in mapping. Consequently, stringent data filtering produces false negatives (missed calls). Here we describe ChIP-exo, where an exonuclease trims ChIP DNA to a precise distance from the crosslinking site. Bound locations are detectable as peak pairs by deep sequencing. Contaminating DNA is degraded or fails to form complementary peak pairs. With the single bp accuracy provided by ChIP-exo, we show an unprecedented view into genome-wide binding of the yeast transcription factors Reb1, Gal4, Phd1, Rap1, and human CTCF. Each of these factors was chosen to address potential limitations of ChIP-exo. We found that binding sites become unambiguous and reveal diverse tendencies governing in vivo DNA-binding specificity that include sequence variants, functionally distinct motifs, motif clustering, secondary interactions, and combinatorial modules within a compound motif.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Gene regulation: Resolving transcription factor binding.Nat Rev Genet. 2011 Dec 29;13(2):71. doi: 10.1038/nrg3153. Nat Rev Genet. 2011. PMID: 22207166 No abstract available.
-
High-resolution chromatin immunoprecipitation.Nat Methods. 2012 Feb;9(2):130. doi: 10.1038/nmeth.1887. Nat Methods. 2012. PMID: 22396966 No abstract available.
References
-
- Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446:572–576. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
