

Role of reactive oxygen and nitrogen species in the vascular responses to inflammation
- PMID: 22154653
- PMCID: PMC3348846
- DOI: 10.1016/j.freeradbiomed.2011.11.002
Role of reactive oxygen and nitrogen species in the vascular responses to inflammation
Abstract
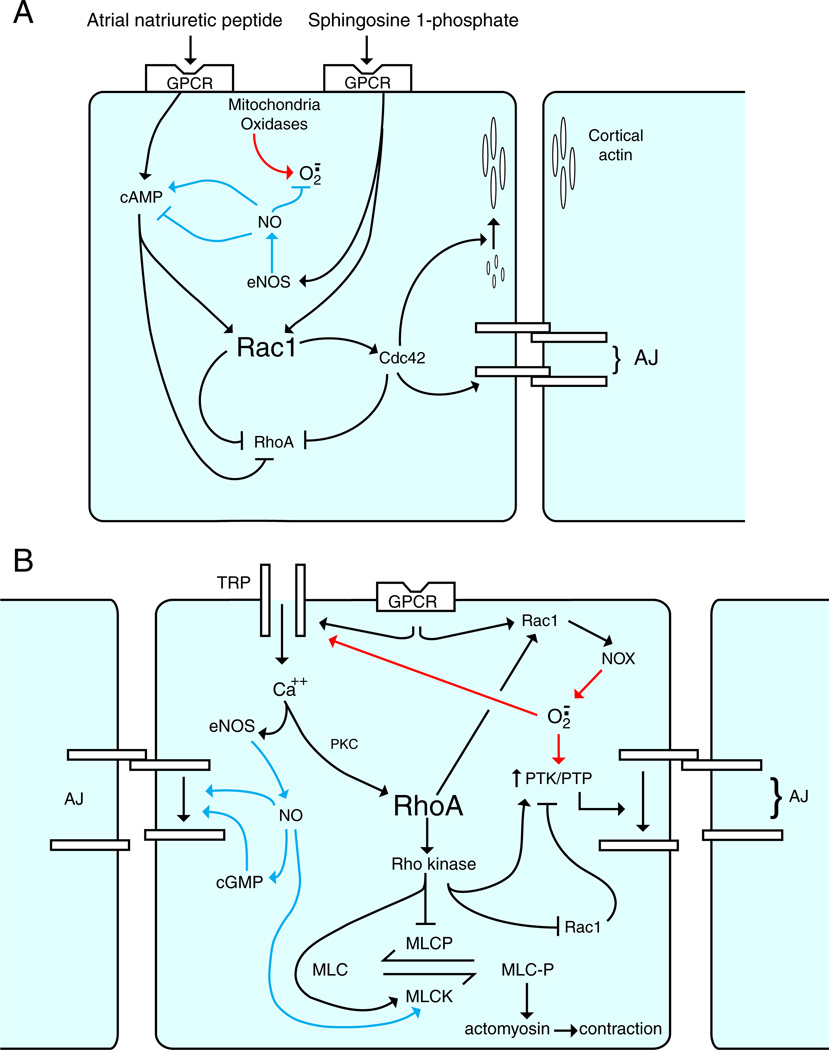
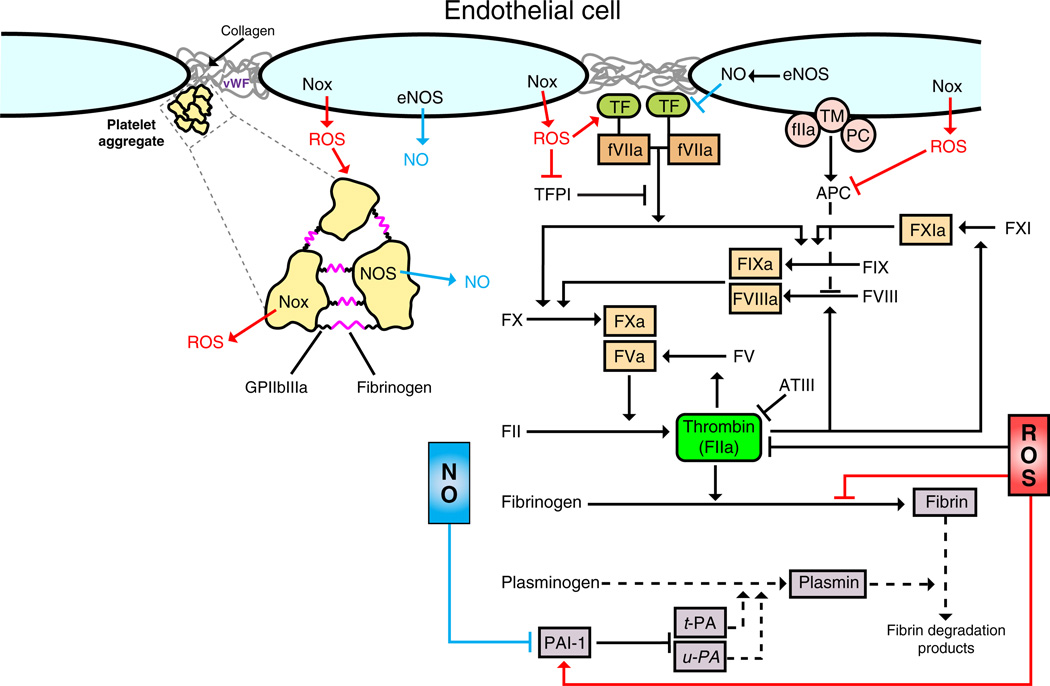
Inflammation is a complex and potentially life-threatening condition that involves the participation of a variety of chemical mediators, signaling pathways, and cell types. The microcirculation, which is critical for the initiation and perpetuation of an inflammatory response, exhibits several characteristic functional and structural changes in response to inflammation. These include vasomotor dysfunction (impaired vessel dilation and constriction), the adhesion and transendothelial migration of leukocytes, endothelial barrier dysfunction (increased vascular permeability), blood vessel proliferation (angiogenesis), and enhanced thrombus formation. These diverse responses of the microvasculature largely reflect the endothelial cell dysfunction that accompanies inflammation and the central role of these cells in modulating processes as varied as blood flow regulation, angiogenesis, and thrombogenesis. The importance of endothelial cells in inflammation-induced vascular dysfunction is also predicated on the ability of these cells to produce and respond to reactive oxygen and nitrogen species. Inflammation seems to upset the balance between nitric oxide and superoxide within (and surrounding) endothelial cells, which is necessary for normal vessel function. This review is focused on defining the molecular targets in the vessel wall that interact with reactive oxygen species and nitric oxide to produce the characteristic functional and structural changes that occur in response to inflammation. This analysis of the literature is consistent with the view that reactive oxygen and nitrogen species contribute significantly to the diverse vascular responses in inflammation and supports efforts that are directed at targeting these highly reactive species to maintain normal vascular health in pathological conditions that are associated with acute or chronic inflammation.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures



References
-
- Granger DN, Senchenkova E. Inflammation and the microcirculation. In: Granger DN, Granger JP, editors. Colloquium Series in Integrated Systems Physiology: from Molecules to Function. Princeton, NJ: Morgan-Claypool Life. Sci; 2010. - PubMed
-
- Ley K. The microcirculation in inflammation. In: Tuma RDW, Ley K, editors. Handbook of Physiology: Microcirculation. San Diego: Academic Press; 2008. pp. 387–448.
-
- Esmon C. Crosstalk between inflammation and thrombosis. Maturitas. 2004;47:305–314. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
