

A persistent stress response to impeded axonal transport leads to accumulation of amyloid-β in the endoplasmic reticulum, and is a probable cause of sporadic Alzheimer's disease
- PMID: 22156573
- PMCID: PMC3363352
- DOI: 10.1159/000332815
A persistent stress response to impeded axonal transport leads to accumulation of amyloid-β in the endoplasmic reticulum, and is a probable cause of sporadic Alzheimer's disease
Abstract
Background and objective: Could a normal--but persistent--stress response to impeded axonal transport lead to late-onset Alzheimer's disease (AD)? Our results offer an affirmative answer, suggesting a mechanism for the abnormal production of amyloid-β (Aβ), triggered by the slowed axonal transport at old age. We hypothesize that Aβ precursor protein (APP) is a sensor at the endoplasmic reticulum (ER) that detects, and signals to the nucleus, abnormalities in axonal transport. When persistently activated, due to chronically slowed-down transport, this signaling pathway leads to accumulation of Aβ within the ER.
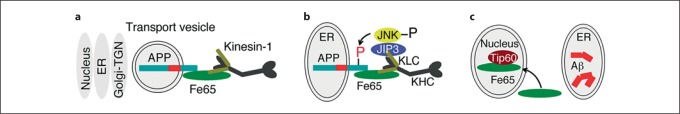
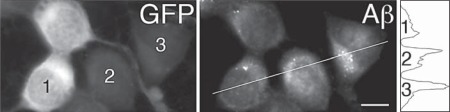
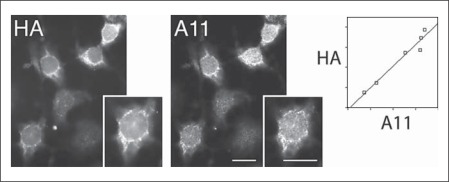
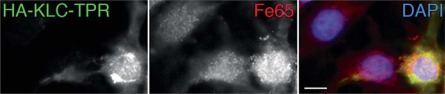
Methods and results: We tested this hypothesis with the neuronal cell line CAD. We show that, normally, a fraction of APP is transported into neurites by recruiting kinesin-1 via the adaptor protein, Fe65. Under conditions that block kinesin-1-dependent transport, APP, Fe65 and kinesin-1 accumulate in the soma, and form a complex at the ER. This complex recruits active c-Jun N-terminal kinase (JNK), which phosphorylates APP at Thr(668). This phosphorylation increases the cleavage of APP by the amyloidogenic pathway, which generates Aβ within the ER lumen, and releases Fe65 into the cytoplasm. Part of the released Fe65 translocates into the nucleus, likely to initiate a gene transcription response to arrested transport. Prolonged arrest of kinesin-1-dependent transport could thus lead to accumulation and oligomerization of Aβ in the ER.
Conclusion: These results support a model where the APP:Fe65 complex is a sensor at the ER for detecting the increased level of kinesin-1 caused by halted transport, which signals to the nucleus, while concomitantly generating an oligomerization-prone pool of Aβ in the ER. Our hypothesis could thus explain a pathogenic mechanism in AD.
Copyright © 2011 S. Karger AG, Basel.
Figures
References
-
- Muresan Z, Muresan V. A phosphorylated, carboxy-terminal fragment of beta-amyloid precursor protein localizes to the splicing factor compartment. Hum Mol Genet. 2004;13:475–488. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
