

A critical role for the PAR-1/MARK-tau axis in mediating the toxic effects of Aβ on synapses and dendritic spines
- PMID: 22156579
- PMCID: PMC3284124
- DOI: 10.1093/hmg/ddr576
A critical role for the PAR-1/MARK-tau axis in mediating the toxic effects of Aβ on synapses and dendritic spines
Abstract
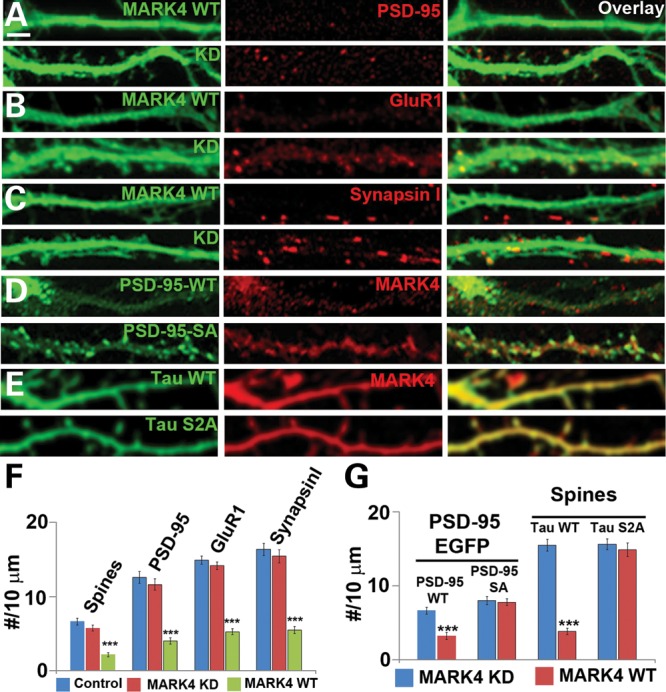
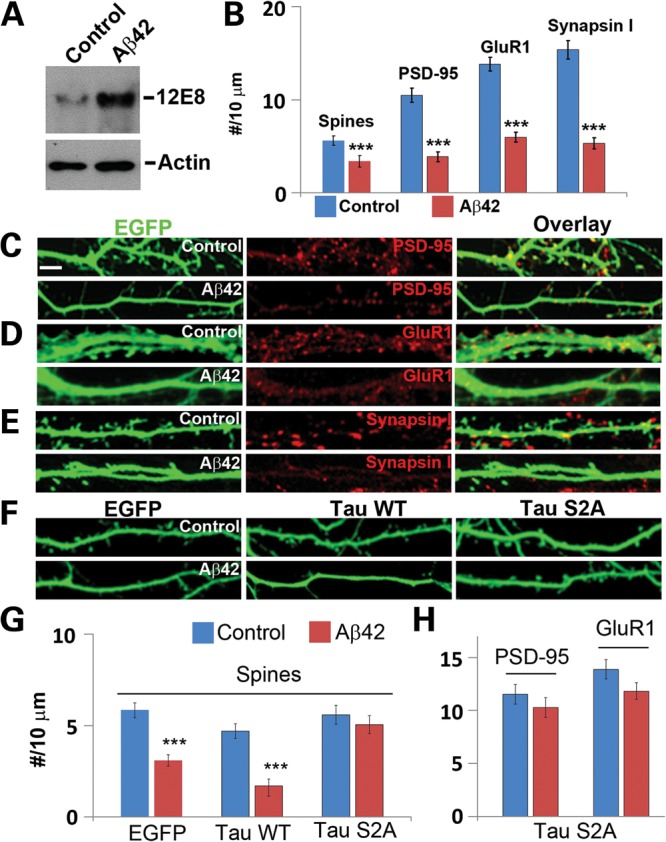
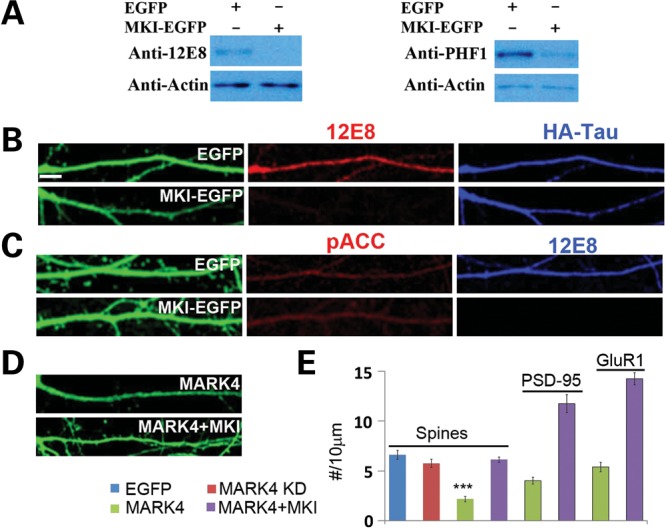
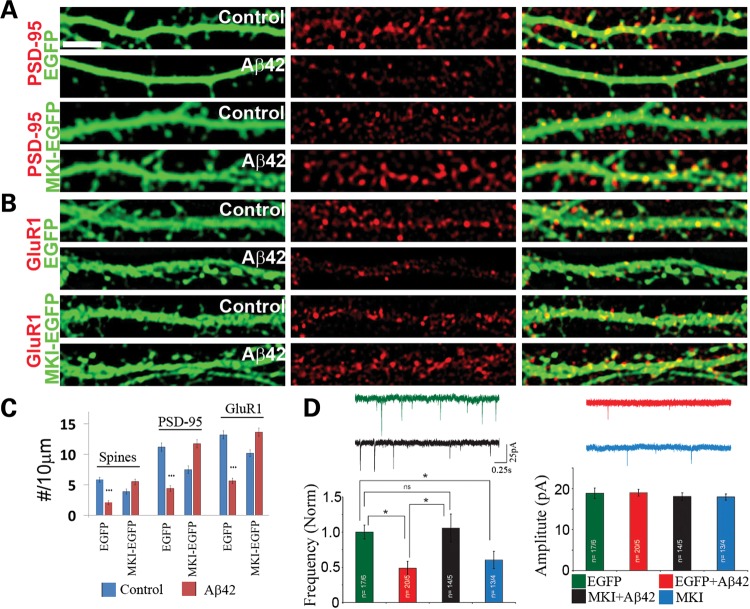
Alzheimer's disease (AD) is the most common neurodegenerative disease and the leading cause of dementia in the elderly. Accumulating evidence supports soluble amyloid-β (Aβ) oligomers as the leading candidate for the causative agent in AD and synapses as the primary site of Aβ oligomer action. However, the molecular and cellular mechanisms by which Aβ oligomers cause synaptic dysfunction and cognitive impairments remain poorly understood. Using primary cultures of rat hippocampal neurons as a model system, we show that the partitioning defective-1 (PAR-1)/microtubule affinity-regulating kinase (MARK) family kinases act as critical mediators of Aβ toxicity on synapses and dendritic spines. Overexpression of MARK4 led to tau hyperphosphorylation, reduced expression of synaptic markers, and loss of dendritic spines and synapses, phenotypes also observed after Aβ treatment. Importantly, expression of a non-phosphorylatable form of tau with the PAR-1/MARK site mutated blocked the synaptic toxicity induced by MARK4 overexpression or Aβ treatment. To probe the involvement of endogenous MARK kinases in mediating the synaptic toxicity of Aβ, we employed a peptide inhibitor capable of effectively and specifically inhibiting the activities of all PAR-1/MARK family members. This inhibitor abrogated the toxic effects of Aβ oligomers on dendritic spines and synapses as assayed at the morphological and electrophysiological levels. Our results reveal a critical role for PAR-1/MARK kinases in AD pathogenesis and suggest PAR-1/MARK inhibitors as potential therapeutics for AD and possibly other tauopathies where aberrant tau hyperphosphorylation is involved.
Figures
References
-
- Selkoe D.J. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
