

Evidence of a robust resident bacteriophage population revealed through analysis of the human salivary virome
- PMID: 22158393
- PMCID: PMC3329113
- DOI: 10.1038/ismej.2011.169
Evidence of a robust resident bacteriophage population revealed through analysis of the human salivary virome
Abstract
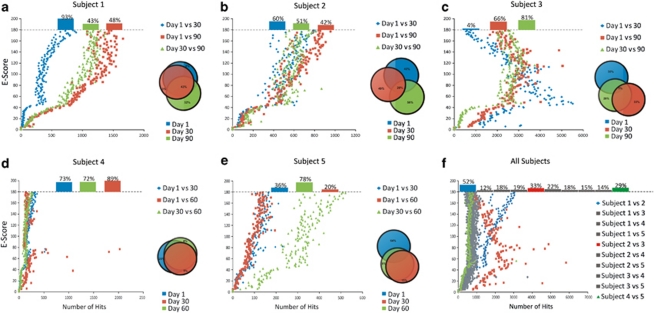
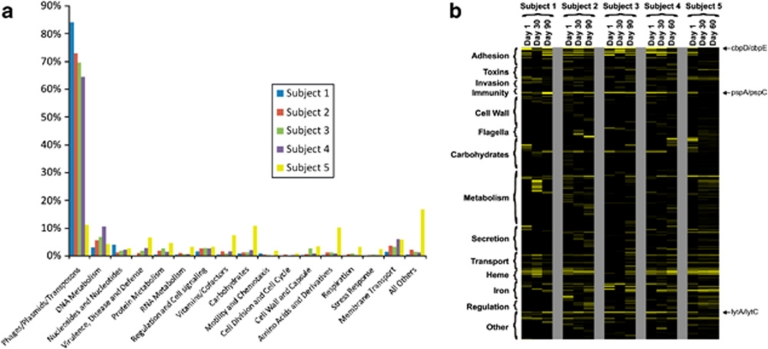
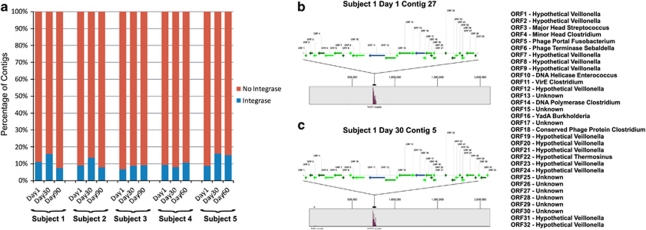
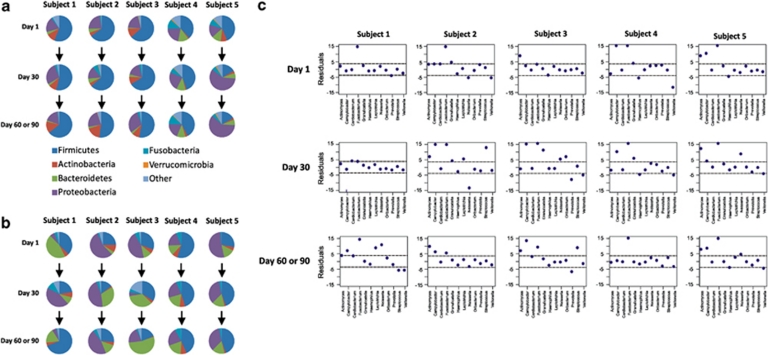
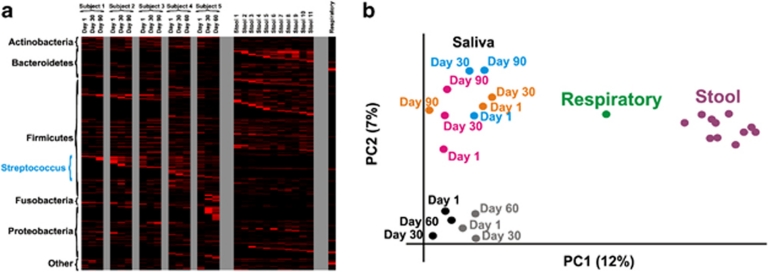
Viruses are the most abundant known infectious agents on the planet and are significant drivers of diversity in a variety of ecosystems. Although there have been numerous studies of viral communities, few have focused on viruses within the indigenous human microbiota. We analyzed 2 267 695 virome reads from viral particles and compared them with 263 516 bacterial 16S rRNA gene sequences from the saliva of five healthy human subjects over a 2- to 3-month period, in order to improve our understanding of the role viruses have in the complex oral ecosystem. Our data reveal viral communities in human saliva dominated by bacteriophages whose constituents are temporally distinct. The preponderance of shared homologs between the salivary viral communities in two unrelated subjects in the same household suggests that environmental factors are determinants of community membership. When comparing salivary viromes to those from human stool and the respiratory tract, each group was distinct, further indicating that habitat is of substantial importance in shaping human viromes. Compared with coexisting bacteria, there was concordance among certain predicted host-virus pairings such as Veillonella and Streptococcus, whereas there was discordance among others such as Actinomyces. We identified 122 728 virulence factor homologs, suggesting that salivary viruses may serve as reservoirs for pathogenic gene function in the oral environment. That the vast majority of human oral viruses are bacteriophages whose putative gene function signifies some have a prominent role in lysogeny, suggests these viruses may have an important role in helping shape the microbial diversity in the human oral cavity.
Figures
References
-
- Andersson AF, Banfield JF. Virus population dynamics and acquired virus resistance in natural microbial communities. Science. 2008;320:1047–1050. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
