

Novel molecular therapies for heritable skin disorders
- PMID: 22158553
- PMCID: PMC3572786
- DOI: 10.1038/jid.2011.389
Novel molecular therapies for heritable skin disorders
Abstract
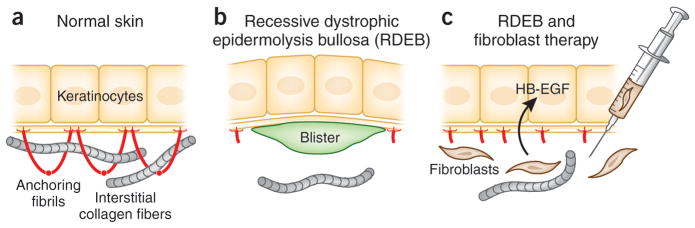
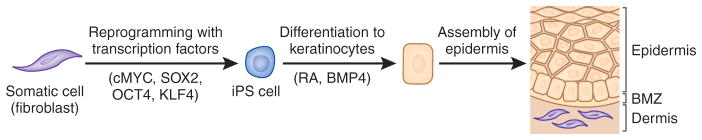
Tremendous progress has been made in the past two decades in molecular genetics of heritable skin diseases, and pathogenic mutations have been identified in as many as 500 distinct human genes. This progress has resulted in improved diagnosis with prognostic implications, has refined genetic counseling, and has formed the basis for prenatal and presymptomatic testing and preimplantation genetic diagnosis. However, there has been relatively little progress in developing effective and specific treatments for these often devastating diseases. However, very recently, a number of novel molecular strategies, including gene therapy, cell-based approaches, and protein replacement therapy, have been explored for the treatment of these conditions. This overview will focus on the prototypic heritable blistering disorders, epidermolysis bullosa, and related keratinopathies, in which significant progress has been made recently toward treatment, and it will illustrate how some of the translational research therapies have already entered the clinical arena.
Figures

References
-
- Atkinson SD, McGilligan VE, Liao H, et al. Development of allele-specific therapeutic siRNA for keratin 5 mutations in epidermolysis bullosa simplex. J Invest Dermatol. 2011 (in press) - PubMed
-
- Aumailley M, Has C, Tunggal L, et al. Molecular basis of inherited skin-blistering disorders and therapeutic implications. Exp Rev Mol Med. 2006;8:1–21. - PubMed
-
- Badiavas EV, Falanga V. Treatment of chronic wounds with bone marrow-derived cells. Arch Dermatol. 2003;139:510–6. - PubMed
-
- Badiavas EV, Abedi M, Butmarc J, et al. Participation of bone marrow derived cells in cutaneous wound healing. J Cell Physiol. 2003;196:245–50. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
