

ATPsite: sequence-based prediction of ATP-binding residues
- PMID: 22165846
- PMCID: PMC3289083
- DOI: 10.1186/1477-5956-9-S1-S4
ATPsite: sequence-based prediction of ATP-binding residues
Abstract
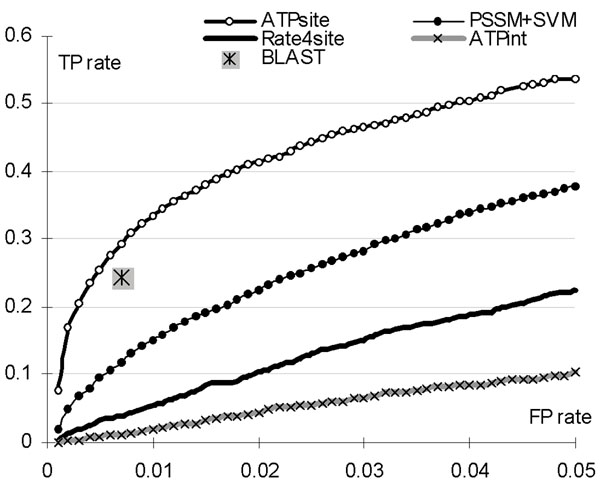
Background: ATP is a ubiquitous nucleotide that provides energy for cellular activities, catalyzes chemical reactions, and is involved in cellular signalling. The knowledge of the ATP-protein interactions helps with annotation of protein functions and finds applications in drug design. The sequence to structure annotation gap motivates development of high-throughput sequence-based predictors of the ATP-binding residues. Moreover, our empirical tests show that the only existing predictor, ATPint, is characterized by relatively low predictive quality.
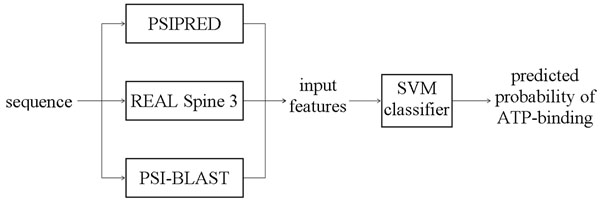
Methods: We propose a novel, high-throughput machine learning-based predictor, ATPsite, which identifies ATP-binding residues from protein sequences. Our predictor utilizes Support Vector Machine classifier and a comprehensive set of input features that are based on the sequence, evolutionary profiles, and the sequence-predicted structural descriptors including secondary structure, solvent accessibility, and dihedral angles.
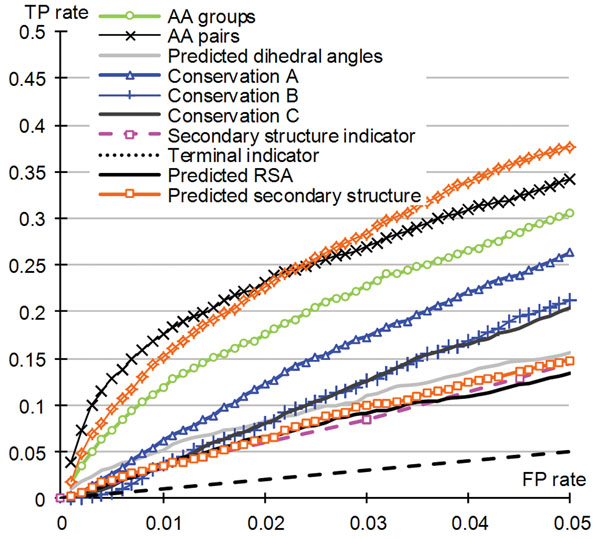
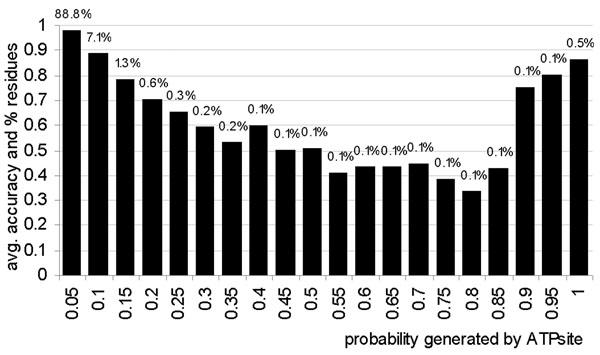
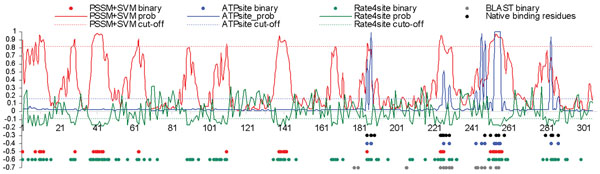
Results: The ATPsite achieves significantly higher Mathews Correlation Coefficient (MCC) and Area Under the ROC Curve (AUC) values when compared with the existing methods including the ATPint, conservation-based rate4site, and alignment-based BLAST predictors. We also assessed the effectiveness of individual input types. The PSSM profile, the conservation scores, and certain features based on amino acid groups are shown to be more effective in predicting the ATP-binding residues than the remaining feature groups.
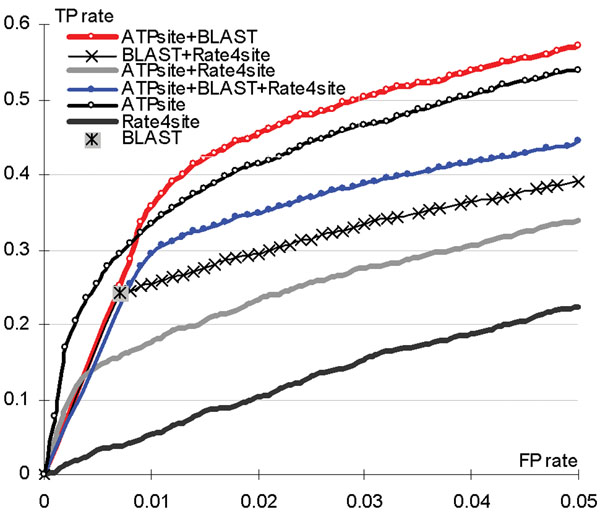
Conclusions: Statistical tests show that ATPsite significantly outperforms existing solutions. The consensus of the ATPsite with the sequence-alignment based predictor is shown to give further improvements.
Figures
References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
