

Staphylococcus aureus DinG, a helicase that has evolved into a nuclease
- PMID: 22166102
- PMCID: PMC3270479
- DOI: 10.1042/BJ20111903
Staphylococcus aureus DinG, a helicase that has evolved into a nuclease
Abstract
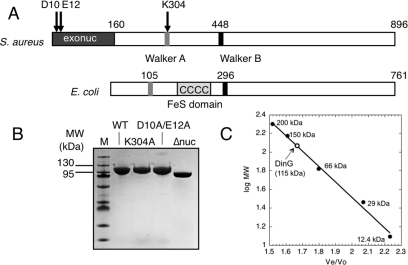
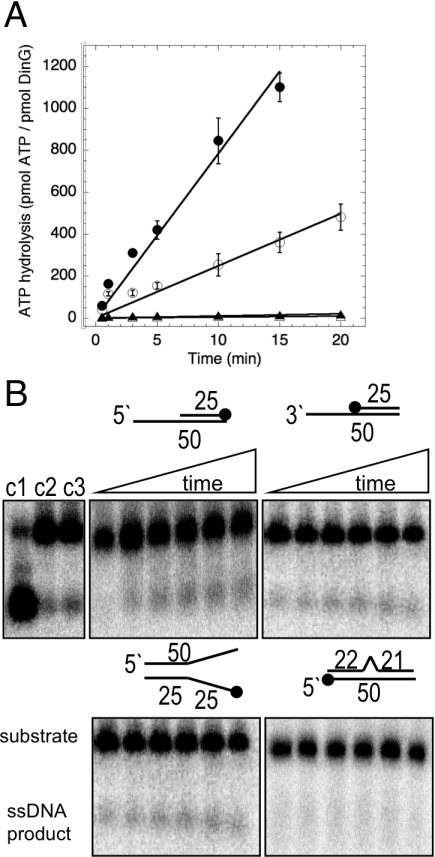
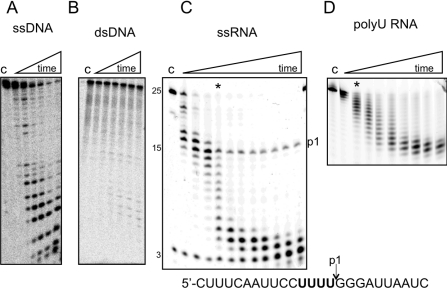
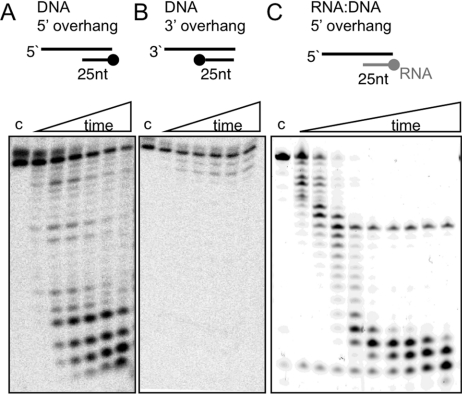
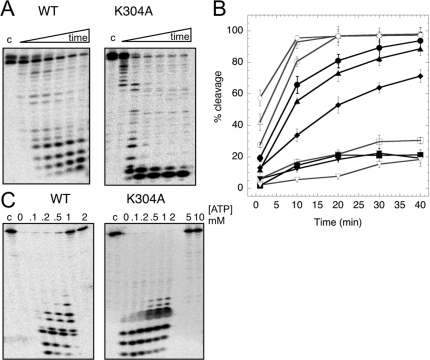
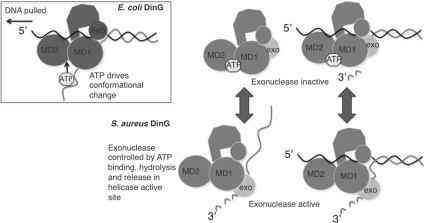
DinG (damage inducible gene G) is a bacterial superfamily 2 helicase with 5′→3′ polarity. DinG is related to the XPD (xeroderma pigmentosum complementation group D) helicase family, and they have in common an FeS (iron–sulfur)-binding domain that is essential for the helicase activity. In the bacilli and clostridia, the DinG helicase has become fused with an N-terminal domain that is predicted to be an exonuclease. In the present paper we show that the DinG protein from Staphylococcus aureus lacks an FeS domain and is not a DNA helicase, although it retains DNA-dependent ATP hydrolysis activity. Instead, the enzyme is an active 3′→5′ exonuclease acting on single-stranded DNA and RNA substrates. The nuclease activity can be modulated by mutation of the ATP-binding cleft of the helicase domain, and is inhibited by ATP or ADP, suggesting a modified role for the inactive helicase domain in the control of the nuclease activity. By degrading rather than displacing RNA or DNA strands, the S. aureus DinG nuclease may accomplish the same function as the canonical DinG helicase.
Figures
References
-
- Singleton M. R., Dillingham M. S., Wigley D. B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007;76:23–50. - PubMed
-
- Gorbalenya A. E., Koonin E. V. Helicases: amino acid sequence comparisons and structure–function relationships. Curr. Opin. Struct. Biol. 1993;3:419–429.
-
- Winkler G. S., Araujo S. J., Fiedler U., Vermeulen W., Coin F., Egly J. M., Hoeijmakers J. H., Wood R. D., Timmers H. T., Weeda G. TFIIH with inactive XPD helicase functions in transcription initiation but is defective in DNA repair. J. Biol. Chem. 2000;275:4258–4266. - PubMed
-
- Sung P., Guzder S. N., Prakash L., Prakash S. Reconstitution of TFIIH and requirement of its DNA helicase subunits, rad3 and rad25, in the incision step of nucleotide excision-repair. J. Biol. Chem. 1996;271:10821–10826. - PubMed
-
- Pugh R. A., Honda M., Leesley H., Thomas A., Lin Y., Nilges M. J., Cann I. K., Spies M. The iron-containing domain is essential in Rad3 helicases for coupling of ATP hydrolysis to DNA translocation and for targeting the helicase to the single-stranded DNA-double-stranded DNA junction. J. Biol. Chem. 2008;283:1732–1743. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous