

New developments on the cheminformatics open workflow environment CDK-Taverna
- PMID: 22166170
- PMCID: PMC3292505
- DOI: 10.1186/1758-2946-3-54
New developments on the cheminformatics open workflow environment CDK-Taverna
Abstract
Background: The computational processing and analysis of small molecules is at heart of cheminformatics and structural bioinformatics and their application in e.g. metabolomics or drug discovery. Pipelining or workflow tools allow for the Lego™-like, graphical assembly of I/O modules and algorithms into a complex workflow which can be easily deployed, modified and tested without the hassle of implementing it into a monolithic application. The CDK-Taverna project aims at building a free open-source cheminformatics pipelining solution through combination of different open-source projects such as Taverna, the Chemistry Development Kit (CDK) or the Waikato Environment for Knowledge Analysis (WEKA). A first integrated version 1.0 of CDK-Taverna was recently released to the public.
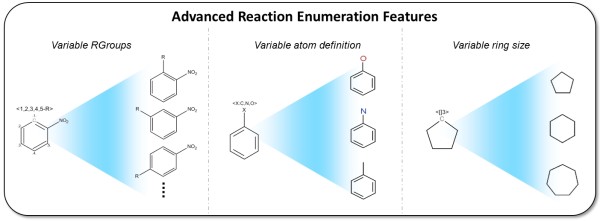
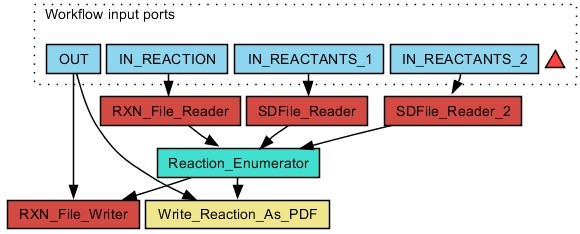
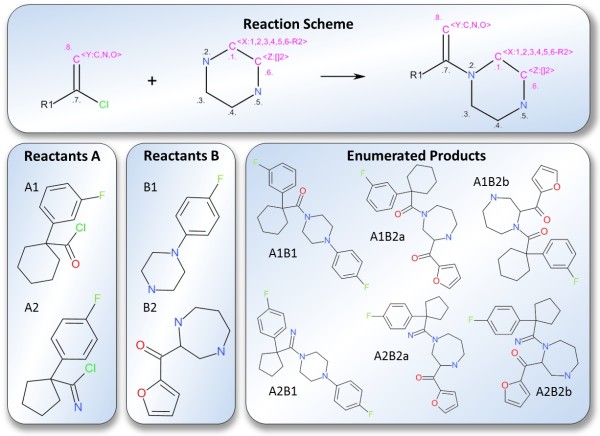
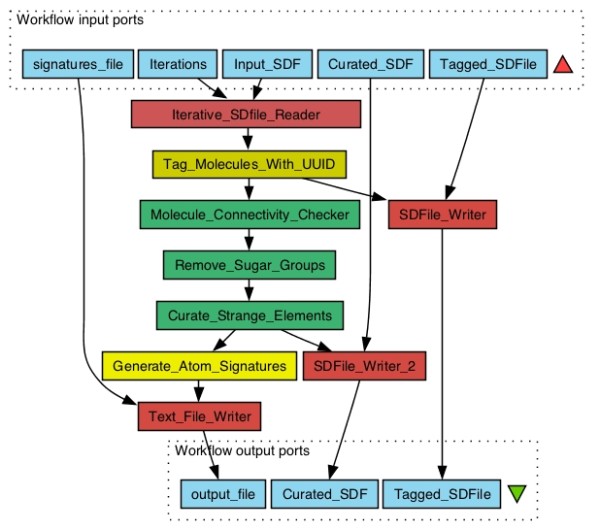
Results: The CDK-Taverna project was migrated to the most up-to-date versions of its foundational software libraries with a complete re-engineering of its worker's architecture (version 2.0). 64-bit computing and multi-core usage by paralleled threads are now supported to allow for fast in-memory processing and analysis of large sets of molecules. Earlier deficiencies like workarounds for iterative data reading are removed. The combinatorial chemistry related reaction enumeration features are considerably enhanced. Additional functionality for calculating a natural product likeness score for small molecules is implemented to identify possible drug candidates. Finally the data analysis capabilities are extended with new workers that provide access to the open-source WEKA library for clustering and machine learning as well as training and test set partitioning. The new features are outlined with usage scenarios.
Conclusions: CDK-Taverna 2.0 as an open-source cheminformatics workflow solution matured to become a freely available and increasingly powerful tool for the biosciences. The combination of the new CDK-Taverna worker family with the already available workflows developed by a lively Taverna community and published on myexperiment.org enables molecular scientists to quickly calculate, process and analyse molecular data as typically found in e.g. today's systems biology scenarios.
Figures






Similar articles
-
The Taverna workflow suite: designing and executing workflows of Web Services on the desktop, web or in the cloud.Nucleic Acids Res. 2013 Jul;41(Web Server issue):W557-61. doi: 10.1093/nar/gkt328. Epub 2013 May 2. Nucleic Acids Res. 2013. PMID: 23640334 Free PMC article.
-
CDK-Taverna: an open workflow environment for cheminformatics.BMC Bioinformatics. 2010 Mar 29;11:159. doi: 10.1186/1471-2105-11-159. BMC Bioinformatics. 2010. PMID: 20346188 Free PMC article.
-
KNIME-CDK: Workflow-driven cheminformatics.BMC Bioinformatics. 2013 Aug 22;14:257. doi: 10.1186/1471-2105-14-257. BMC Bioinformatics. 2013. PMID: 24103053 Free PMC article.
-
Cheminformatics analysis and learning in a data pipelining environment.Mol Divers. 2006 Aug;10(3):283-99. doi: 10.1007/s11030-006-9041-5. Epub 2006 Sep 22. Mol Divers. 2006. PMID: 17031533 Review.
-
Web tools for predictive toxicology model building.Expert Opin Drug Metab Toxicol. 2012 Jul;8(7):791-801. doi: 10.1517/17425255.2012.685158. Epub 2012 May 12. Expert Opin Drug Metab Toxicol. 2012. PMID: 22577953 Review.
Cited by
-
A survey of quantitative descriptions of molecular structure.Curr Top Med Chem. 2012;12(18):1946-56. doi: 10.2174/156802612804910278. Curr Top Med Chem. 2012. PMID: 23110530 Free PMC article.
-
The Chemistry Development Kit (CDK) v2.0: atom typing, depiction, molecular formulas, and substructure searching.J Cheminform. 2017 Jun 6;9(1):33. doi: 10.1186/s13321-017-0220-4. J Cheminform. 2017. PMID: 29086040 Free PMC article.
-
Applications of the InChI in cheminformatics with the CDK and Bioclipse.J Cheminform. 2013 Mar 13;5(1):14. doi: 10.1186/1758-2946-5-14. J Cheminform. 2013. PMID: 23497723 Free PMC article.
-
The Taverna workflow suite: designing and executing workflows of Web Services on the desktop, web or in the cloud.Nucleic Acids Res. 2013 Jul;41(Web Server issue):W557-61. doi: 10.1093/nar/gkt328. Epub 2013 May 2. Nucleic Acids Res. 2013. PMID: 23640334 Free PMC article.
-
Scientific workflow systems: Pipeline Pilot and KNIME.J Comput Aided Mol Des. 2012 Jul;26(7):801-4. doi: 10.1007/s10822-012-9577-7. Epub 2012 May 27. J Comput Aided Mol Des. 2012. PMID: 22644661 Free PMC article. No abstract available.
References
-
- Shon J, Ohkawa H, Hammer J. Scientific workflows as productivity tools for drug discovery. Current opinion in drug discovery and development. 2008;11(3):381–388. - PubMed
-
- Oinn T, Li P, Kell D, Goble C, Goderis A, Greenwood M, Hull D, Stevens R, Turi D, Zhao J. Taverna/my Grid: Aligning a Workflow System with the Life Sciences Community. Workflows for e-Science. 2007. pp. 300–319.http://www.springerlink.com/index/l9425v576v544vv3.pdf
-
- Missier P, Soiland-Reyes S, Owen S, Tan W, Nenadic A, Dunlop I, Williams A, Oinn T, Goble C. Taverna, Reloaded. Lecture Notes in Computer Science. 2010;6187:471–481. doi: 10.1007/978-3-642-13818-8_33. - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
