

The chemokine CCL2 increases Nav1.8 sodium channel activity in primary sensory neurons through a Gβγ-dependent mechanism
- PMID: 22171040
- PMCID: PMC6623900
- DOI: 10.1523/JNEUROSCI.3386-11.2011
The chemokine CCL2 increases Nav1.8 sodium channel activity in primary sensory neurons through a Gβγ-dependent mechanism
Abstract
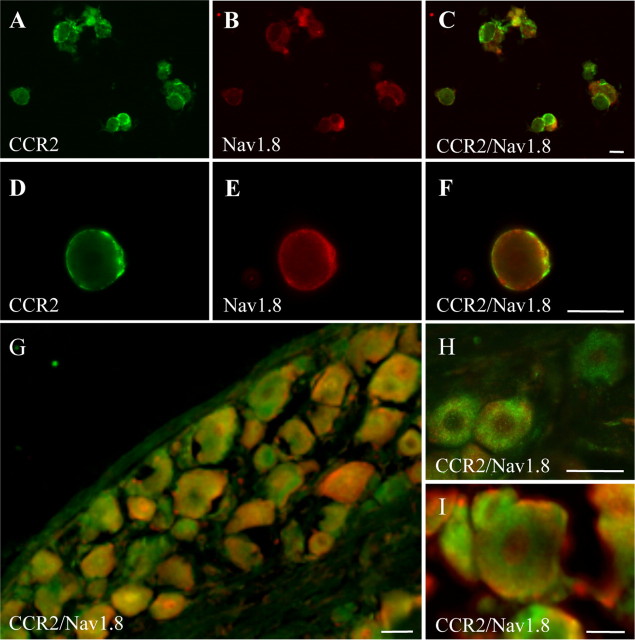
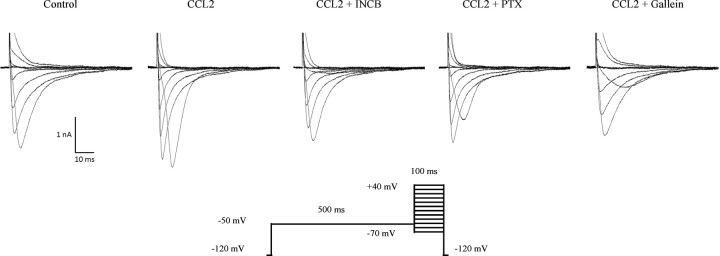
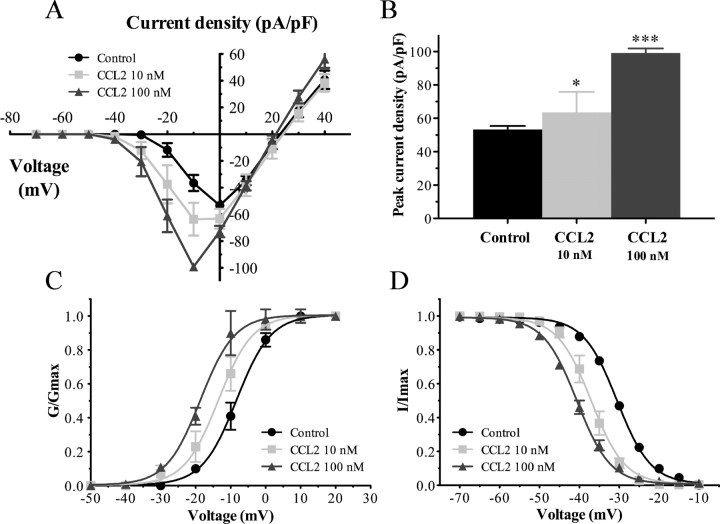
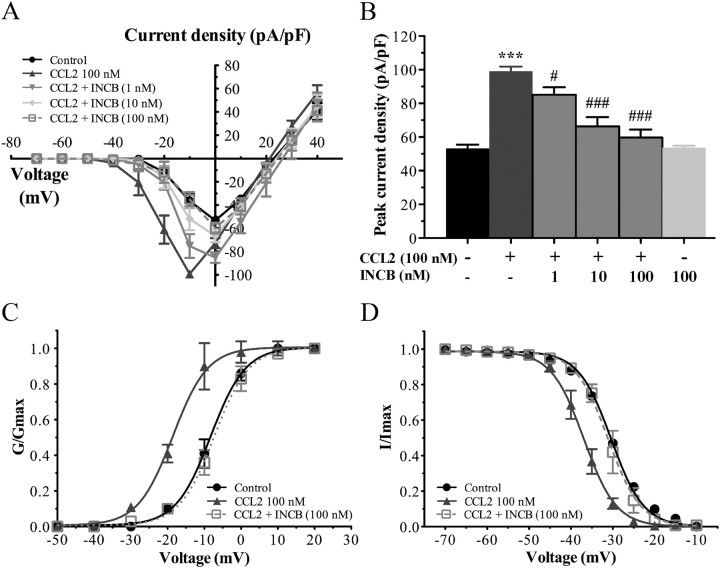
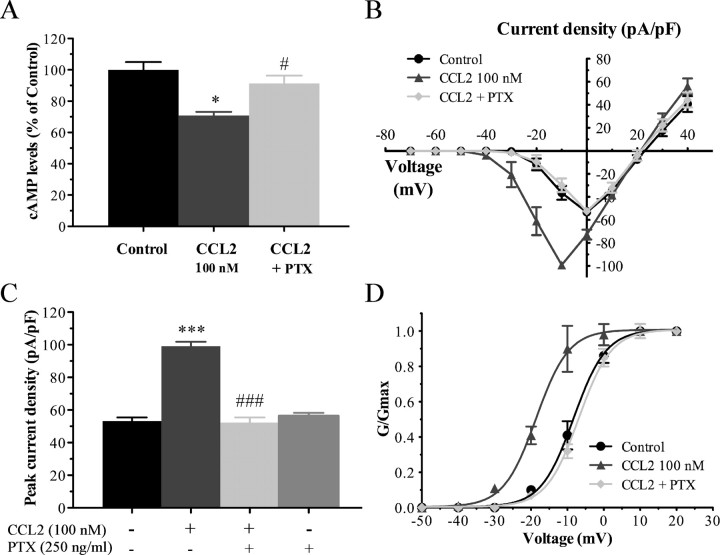
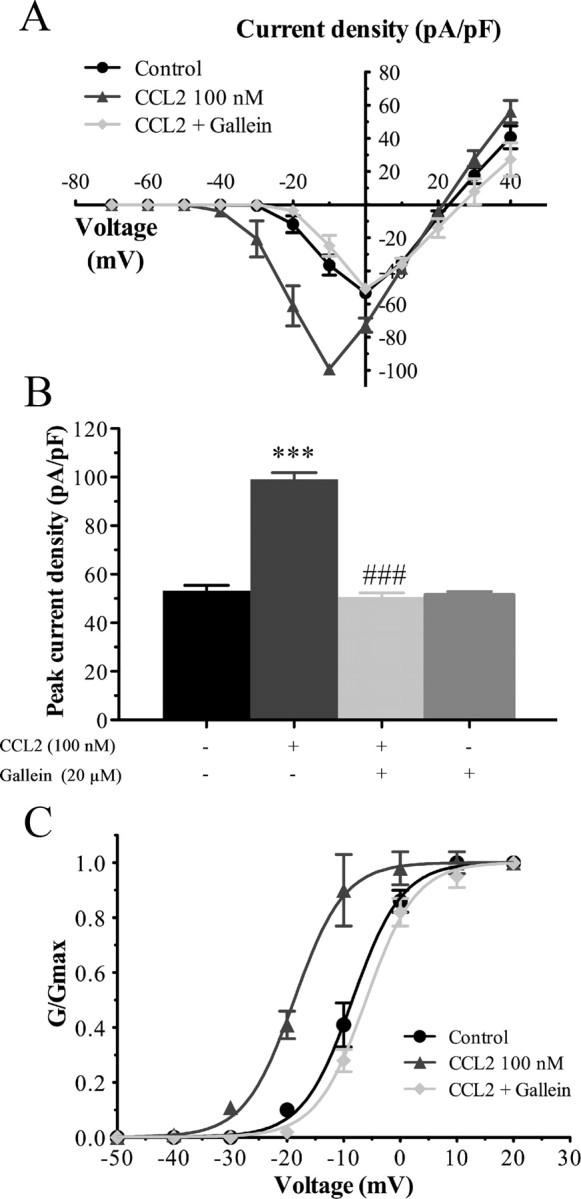
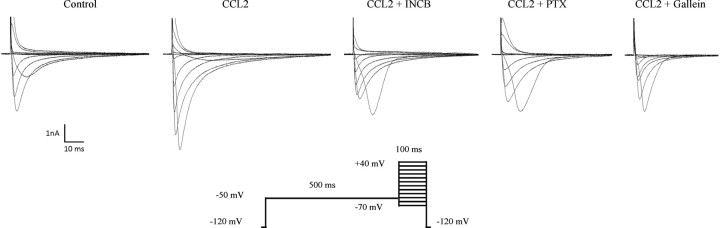
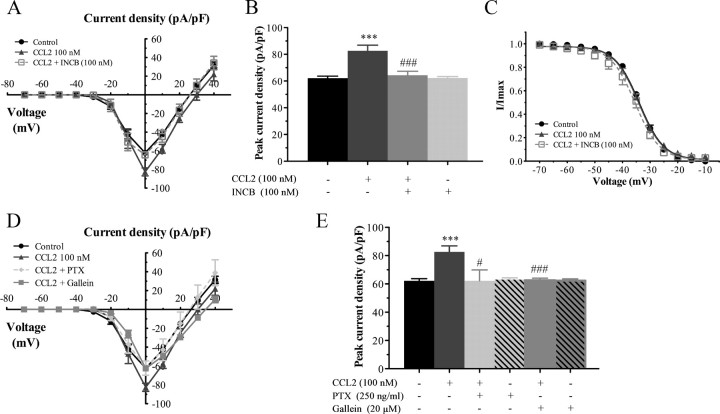
Changes in function of voltage-gated sodium channels in nociceptive primary sensory neurons participate in the development of peripheral hyperexcitability that occurs in neuropathic and inflammatory chronic pain conditions. Among them, the tetrodotoxin-resistant (TTX-R) sodium channel Na(v)1.8, primarily expressed by small- and medium-sized dorsal root ganglion (DRG) neurons, substantially contributes to the upstroke of action potential in these neurons. Compelling evidence also revealed that the chemokine CCL2 plays a critical role in chronic pain facilitation via its binding to CCR2 receptors. In this study, we therefore investigated the effects of CCL2 on the density and kinetic properties of TTX-R Na(v)1.8 currents in acutely small/medium dissociated lumbar DRG neurons from naive adult rats. Whole-cell patch-clamp recordings demonstrated that CCL2 concentration-dependently increased TTX-resistant Na(v)1.8 current densities in both small- and medium-diameter sensory neurons. Incubation with CCL2 also shifted the activation and steady-state inactivation curves of Na(v)1.8 in a hyperpolarizing direction in small sensory neurons. No change in the activation and inactivation kinetics was, however, observed in medium-sized nociceptive neurons. Our electrophysiological recordings also demonstrated that the selective CCR2 antagonist INCB3344 [N-[2-[[(3S,4S)-1-E4-(1,3-benzodioxol-5-yl)-4-hydroxycyclohexyl]-4-ethoxy-3-pyrrolidinyl]amino]-2-oxoethyl]-3-(trifluoromethyl)benzamide] blocks the potentiation of Na(v)1.8 currents by CCL2 in a concentration-dependent manner. Furthermore, the enhancement in Na(v)1.8 currents was prevented by pretreatment with pertussis toxin (PTX) or gallein (a Gβγ inhibitor), indicating the involvement of Gβγ released from PTX-sensitive G(i/o)-proteins in the cross talk between CCR2 and Na(v)1.8. Together, our data clearly demonstrate that CCL2 may excite primary sensory neurons by acting on the biophysical properties of Na(v)1.8 currents via a CCR2/Gβγ-dependent mechanism.
Figures
Similar articles
-
CC chemokine ligand 2 upregulates the current density and expression of TRPV1 channels and Nav1.8 sodium channels in dorsal root ganglion neurons.J Neuroinflammation. 2012 Aug 8;9:189. doi: 10.1186/1742-2094-9-189. J Neuroinflammation. 2012. PMID: 22870919 Free PMC article.
-
CC chemokine ligand 2 (CCL2) enhances TTX-sensitive sodium channel activity of primary afferent neurons in the complete Freud adjuvant-induced inflammatory pain model.Acta Biochim Biophys Sin (Shanghai). 2018 Dec 1;50(12):1219-1226. doi: 10.1093/abbs/gmy123. Acta Biochim Biophys Sin (Shanghai). 2018. PMID: 30339176
-
Dexmedetomidine inhibits Tetrodotoxin-resistant Nav1.8 sodium channel activity through Gi/o-dependent pathway in rat dorsal root ganglion neurons.Mol Brain. 2015 Mar 3;8:15. doi: 10.1186/s13041-015-0105-2. Mol Brain. 2015. PMID: 25761941 Free PMC article.
-
Structure and Function of Sodium Channel Nav1.3 in Neurological Disorders.Cell Mol Neurobiol. 2023 Mar;43(2):575-584. doi: 10.1007/s10571-022-01211-w. Epub 2022 Mar 24. Cell Mol Neurobiol. 2023. PMID: 35332400 Free PMC article. Review.
-
Pain disorders and erythromelalgia caused by voltage-gated sodium channel mutations.Curr Neurol Neurosci Rep. 2012 Feb;12(1):76-83. doi: 10.1007/s11910-011-0233-8. Curr Neurol Neurosci Rep. 2012. PMID: 21984269 Review.
Cited by
-
Orthosteric muscarinic receptor activation by the insect repellent IR3535 opens new prospects in insecticide-based vector control.Sci Rep. 2020 Apr 22;10(1):6842. doi: 10.1038/s41598-020-63957-x. Sci Rep. 2020. PMID: 32321987 Free PMC article.
-
CCL2-ethanol interactions and hippocampal synaptic protein expression in a transgenic mouse model.Front Integr Neurosci. 2014 Apr 4;8:29. doi: 10.3389/fnint.2014.00029. eCollection 2014. Front Integr Neurosci. 2014. PMID: 24772072 Free PMC article.
-
CCL2 released at tumoral level contributes to the hyperalgesia evoked by intratibial inoculation of NCTC 2472 but not B16-F10 cells in mice.Naunyn Schmiedebergs Arch Pharmacol. 2012 Nov;385(11):1053-61. doi: 10.1007/s00210-012-0787-2. Epub 2012 Sep 14. Naunyn Schmiedebergs Arch Pharmacol. 2012. PMID: 22976830
-
A pharmacological interactome between COVID-19 patient samples and human sensory neurons reveals potential drivers of neurogenic pulmonary dysfunction.SSRN [Preprint]. 2020 May 4:3581446. doi: 10.2139/ssrn.3581446. SSRN. 2020. Update in: Brain Behav Immun. 2020 Oct;89:559-568. doi: 10.1016/j.bbi.2020.05.078. PMID: 32714114 Free PMC article. Updated. Preprint.
-
Mechanistic insights into the role of the chemokine CCL2/CCR2 axis in dorsal root ganglia to peripheral inflammation and pain hypersensitivity.J Neuroinflammation. 2021 Mar 23;18(1):79. doi: 10.1186/s12974-021-02125-y. J Neuroinflammation. 2021. PMID: 33757529 Free PMC article.
References
-
- Amir R, Argoff CE, Bennett GJ, Cummins TR, Durieux ME, Gerner P, Gold MS, Porreca F, Strichartz GR. The role of sodium channels in chronic inflammatory and neuropathic pain. J Pain. 2006;7:S1–S29. - PubMed
-
- Bajetto A, Bonavia R, Barbero S, Schettini G. Characterization of chemokines and their receptors in the central nervous system: physiopathological implications. J Neurochem. 2002;82:1311–1329. - PubMed
-
- Bear B, Asgian J, Termin A, Zimmermann N. Small molecules targeting sodium and calcium channels for neuropathic pain. Curr Opin Drug Discov Devel. 2009;12:543–561. - PubMed
-
- Bhangoo S, Ren D, Miller RJ, Henry KJ, Lineswala J, Hamdouchi C, Li B, Monahan PE, Chan DM, Ripsch MS, White FA. Delayed functional expression of neuronal chemokine receptors following focal nerve demyelination in the rat: a mechanism for the development of chronic sensitization of peripheral nociceptors. Mol Pain. 2007;3:38. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources