

WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing
- PMID: 22172722
- PMCID: PMC3574979
- DOI: 10.1016/j.ccr.2011.10.016
WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing
Abstract
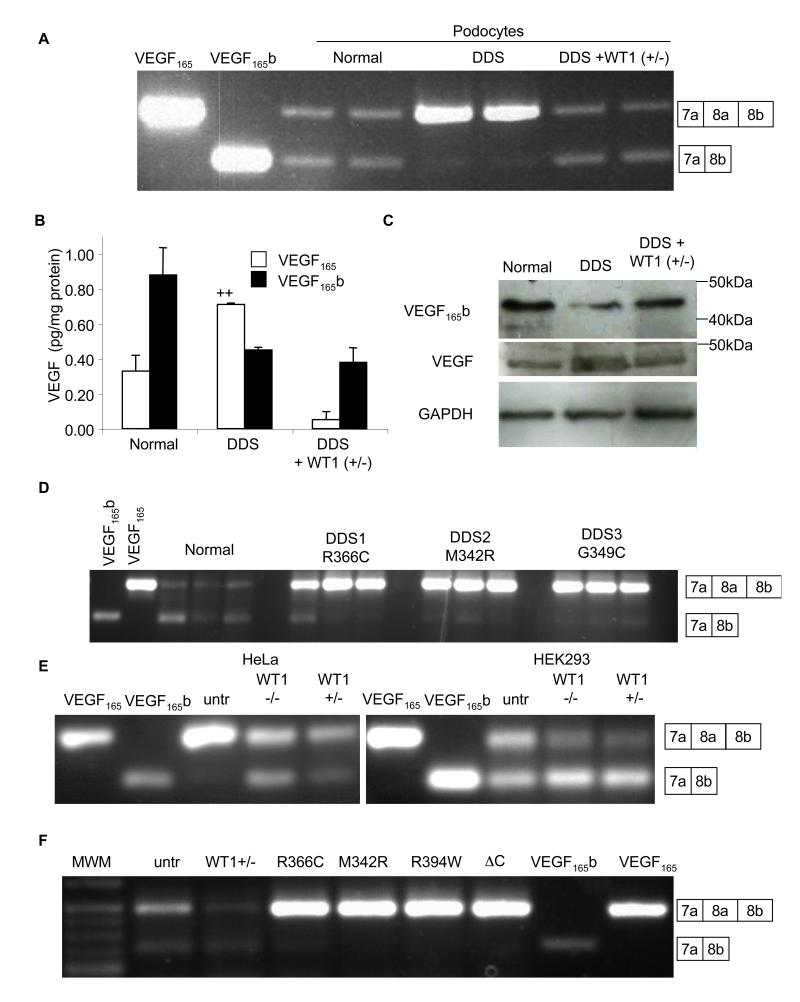
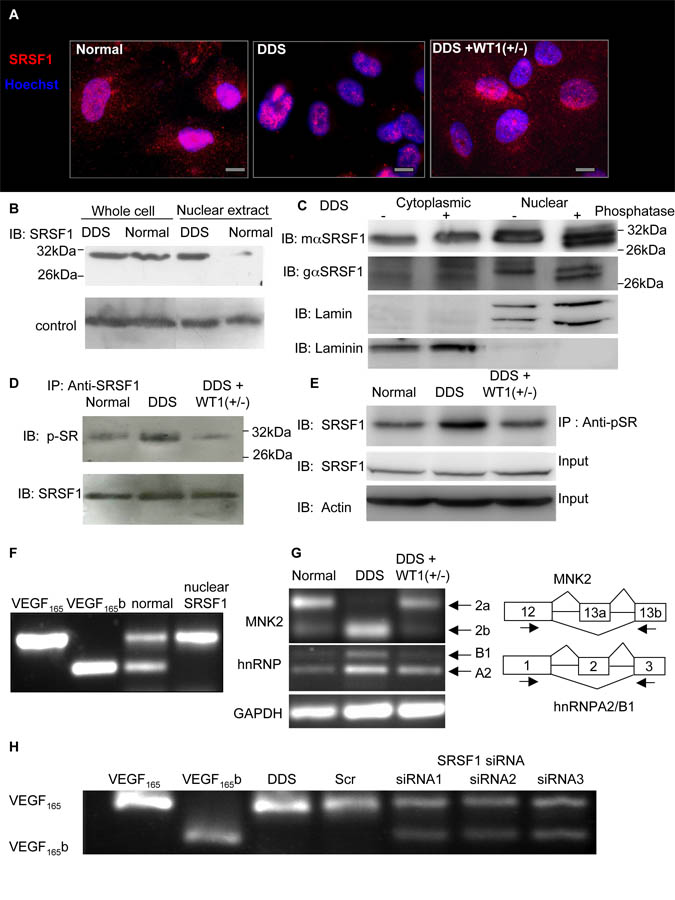
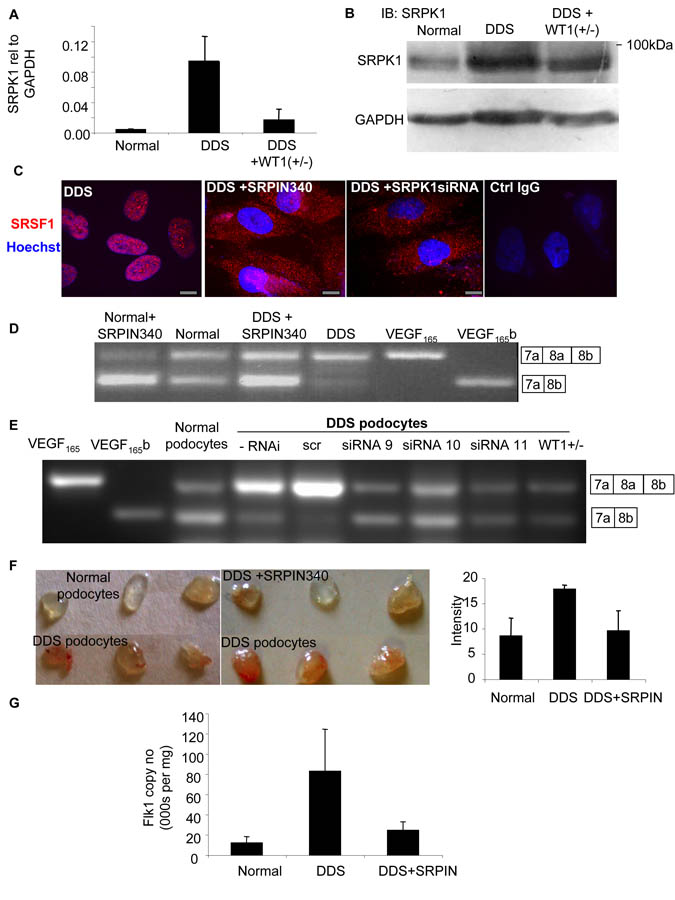
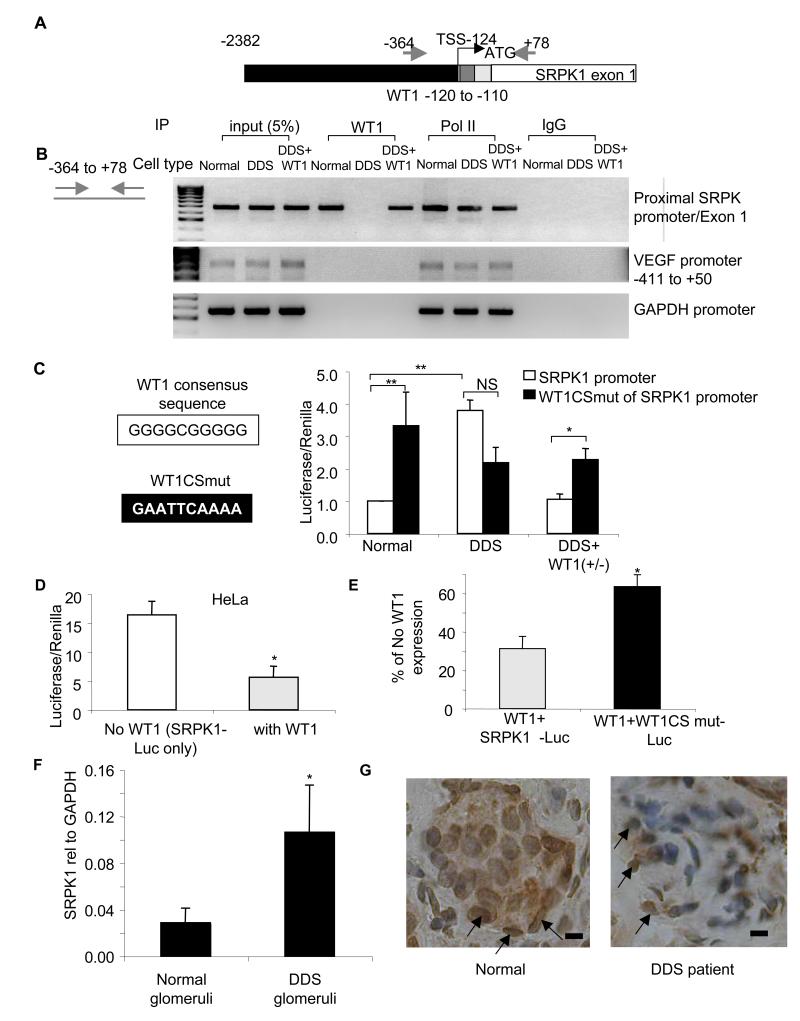
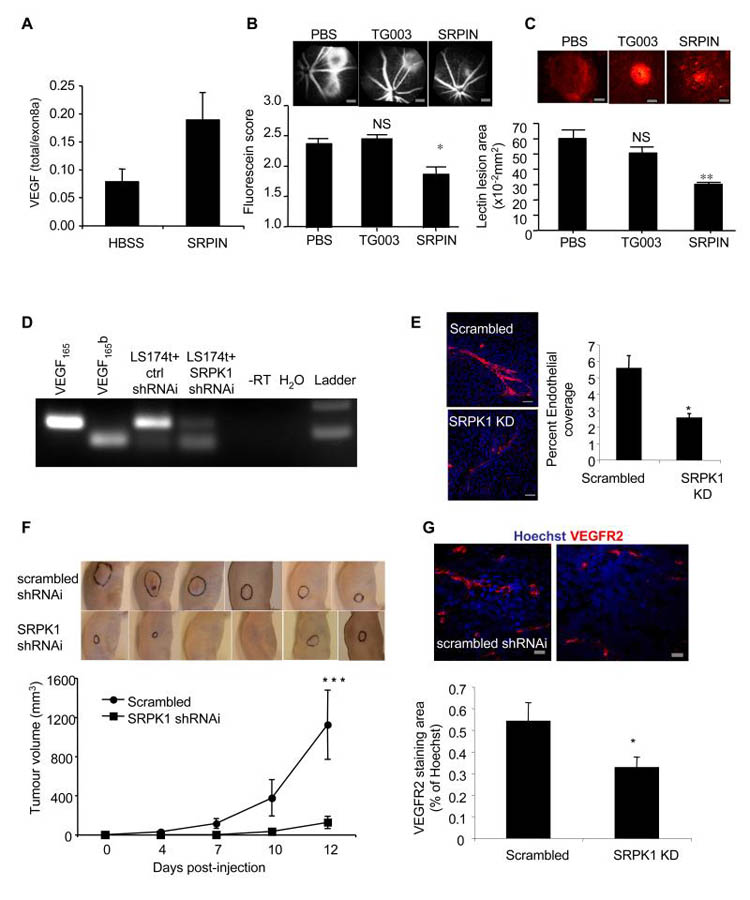
Angiogenesis is regulated by the balance of proangiogenic VEGF(165) and antiangiogenic VEGF(165)b splice isoforms. Mutations in WT1, the Wilms' tumor suppressor gene, suppress VEGF(165)b and cause abnormal gonadogenesis, renal failure, and Wilms' tumors. In WT1 mutant cells, reduced VEGF(165)b was due to lack of WT1-mediated transcriptional repression of the splicing-factor kinase SRPK1. WT1 bound to the SRPK1 promoter, and repressed expression through a specific WT1 binding site. In WT1 mutant cells SRPK1-mediated hyperphosphorylation of the oncogenic RNA binding protein SRSF1 regulated splicing of VEGF and rendered WT1 mutant cells proangiogenic. Altered VEGF splicing was reversed by wild-type WT1, knockdown of SRSF1, or SRPK1 and inhibition of SRPK1, which prevented in vitro and in vivo angiogenesis and associated tumor growth.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Angiogenesis: What's the alternative?Nat Rev Cancer. 2012 Jan 12;12(2):80-1. doi: 10.1038/nrc3207. Nat Rev Cancer. 2012. PMID: 22237396 No abstract available.
References
-
- Artac RA, McFee RM, Smith RA, Baltes-Breitwisch MM, Clopton DT, Cupp AS. Neutralization of vascular endothelial growth factor antiangiogenic isoforms is more effective than treatment with proangiogenic isoforms in stimulating vascular development and follicle progression in the perinatal rat ovary. Biol Reprod. 2009;81:978–988. - PMC - PubMed
-
- Ballmer-Hofer K, Andersson AE, Ratcliffe LE, Berger P. Neuropilin-1 promotes VEGFR-2 trafficking through Rab11 vesicles thereby specifying signal output. Blood. 2011 - PubMed
-
- Bates DO, Cui TG, Doughty JM, Winkler M, Sugiono M, Shields JD, Peat D, Gillatt D, Harper SJ. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002;62:4123–4131. - PubMed
-
- Blann AD, Li JL, Li C, Kumar S. Increased serum VEGF in 13 children with Wilms’ tumour falls after surgery but rising levels predict poor prognosis. Cancer Lett. 2001;173:183–186. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 07-0605/AICR_/Worldwide Cancer Research/United Kingdom
- PG/08/054/25272/BHF_/British Heart Foundation/United Kingdom
- BS/06/005/20340/BHF_/British Heart Foundation/United Kingdom
- PG/11/20/28792/BHF_/British Heart Foundation/United Kingdom
- G10002073/MRC_/Medical Research Council/United Kingdom
- A8451/CRUK_/Cancer Research UK/United Kingdom
- 079736/WT_/Wellcome Trust/United Kingdom
- CF11392/A8451/CRUK_/Cancer Research UK/United Kingdom
- C11392/A10484/CRUK_/Cancer Research UK/United Kingdom
- PG08/022/21636/BHF_/British Heart Foundation/United Kingdom
- GR0600920/MRC_/Medical Research Council/United Kingdom
- A5730/CRUK_/Cancer Research UK/United Kingdom
- P30 EY003040/EY/NEI NIH HHS/United States
- EY03040/EY/NEI NIH HHS/United States
- G0600920/MRC_/Medical Research Council/United Kingdom
- FS06/003/BHF_/British Heart Foundation/United Kingdom
- G0600920(78877)/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
