

Interfacial inhibitors: targeting macromolecular complexes
- PMID: 22173432
- PMCID: PMC7380715
- DOI: 10.1038/nrd3404
Interfacial inhibitors: targeting macromolecular complexes
Erratum in
- Nat Rev Drug Discov. 2012 Mar;11(3):250
Abstract
Interfacial inhibitors belong to a broad class of natural products and synthetic drugs that are commonly used to treat cancers as well as bacterial and HIV infections. They bind selectively to interfaces as macromolecular machines assemble and are set in motion. The bound drugs transiently arrest the targeted molecular machines, which can initiate allosteric effects, or desynchronize macromolecular machines that normally function in concert. Here, we review five archetypical examples of interfacial inhibitors: the camptothecins, etoposide, the quinolone antibiotics, the vinca alkaloids and the novel anti-HIV inhibitor raltegravir. We discuss the common and diverging elements between interfacial and allosteric inhibitors and give a perspective for the rationale and methods used to discover novel interfacial inhibitors.
Conflict of interest statement
Competing interests statement
The authors declare no competing financial interests.
Figures
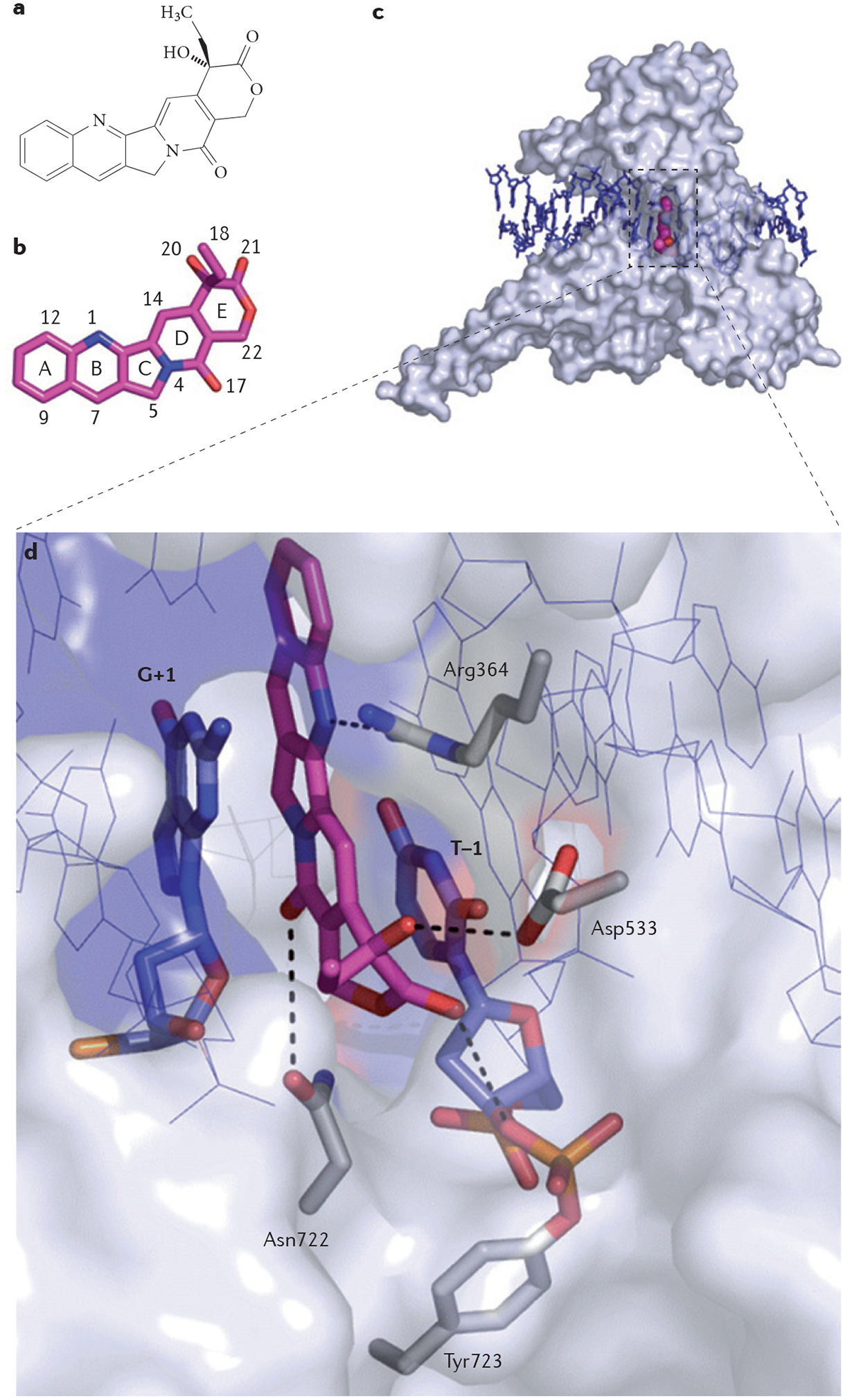
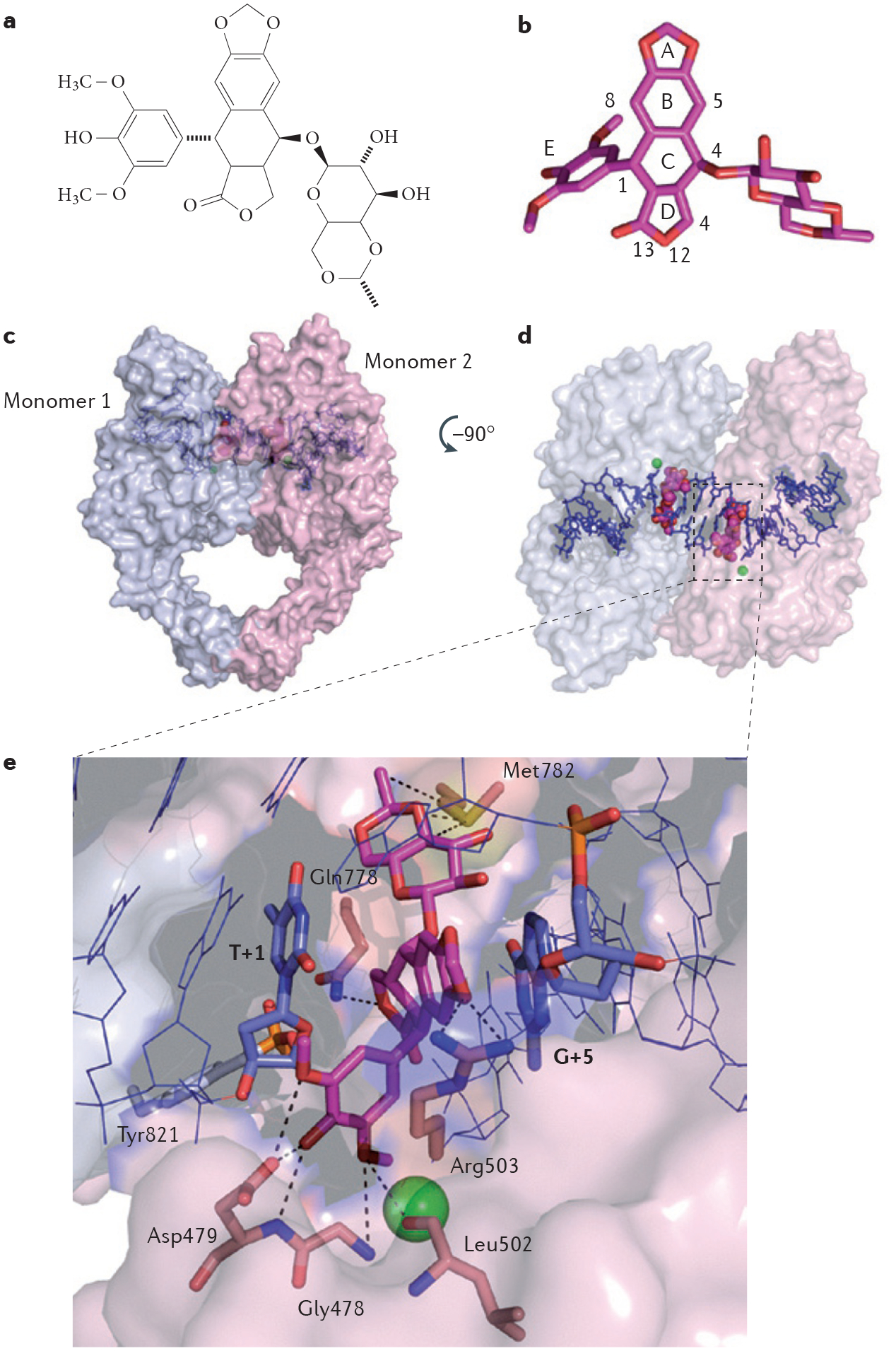
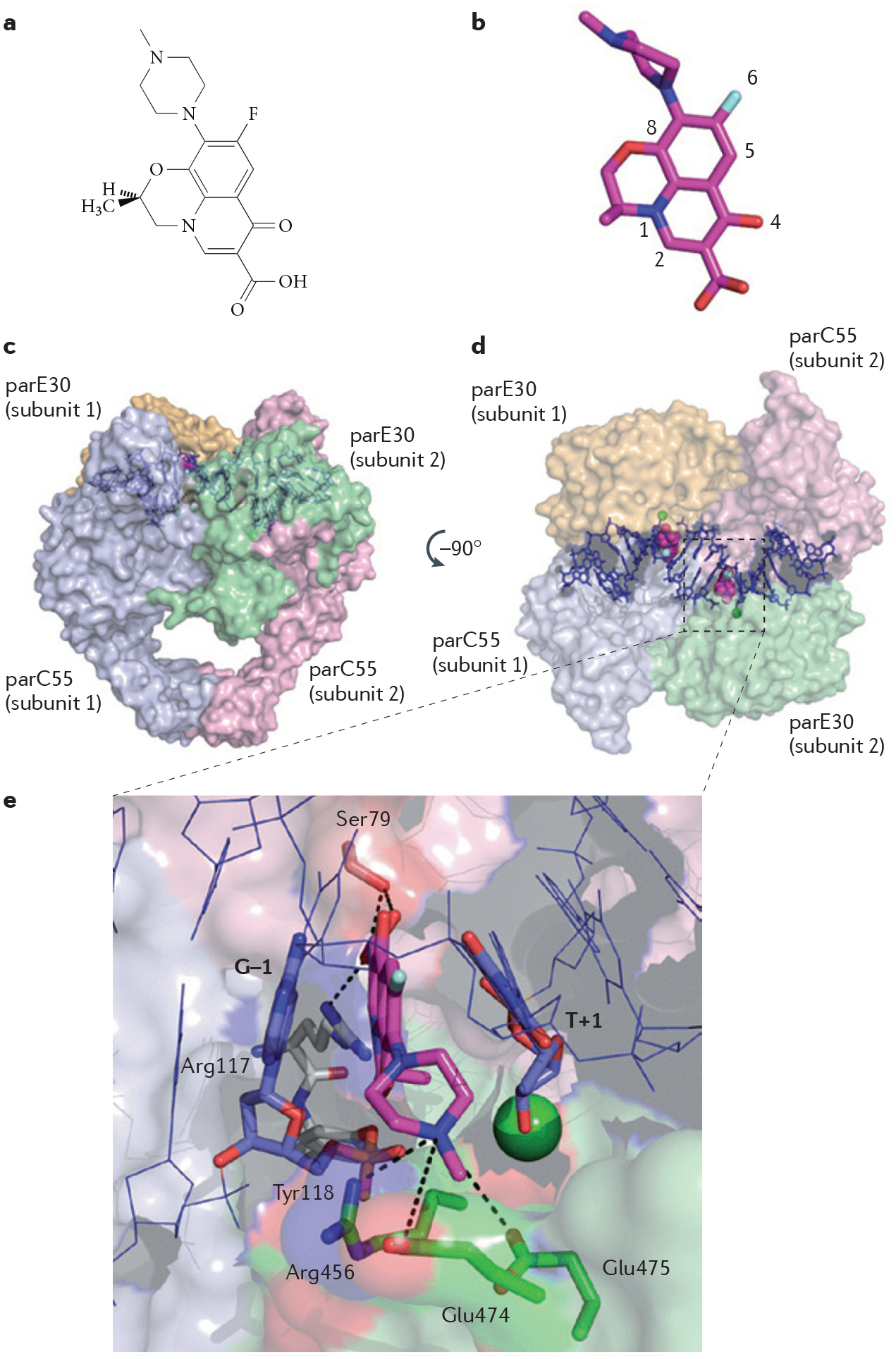
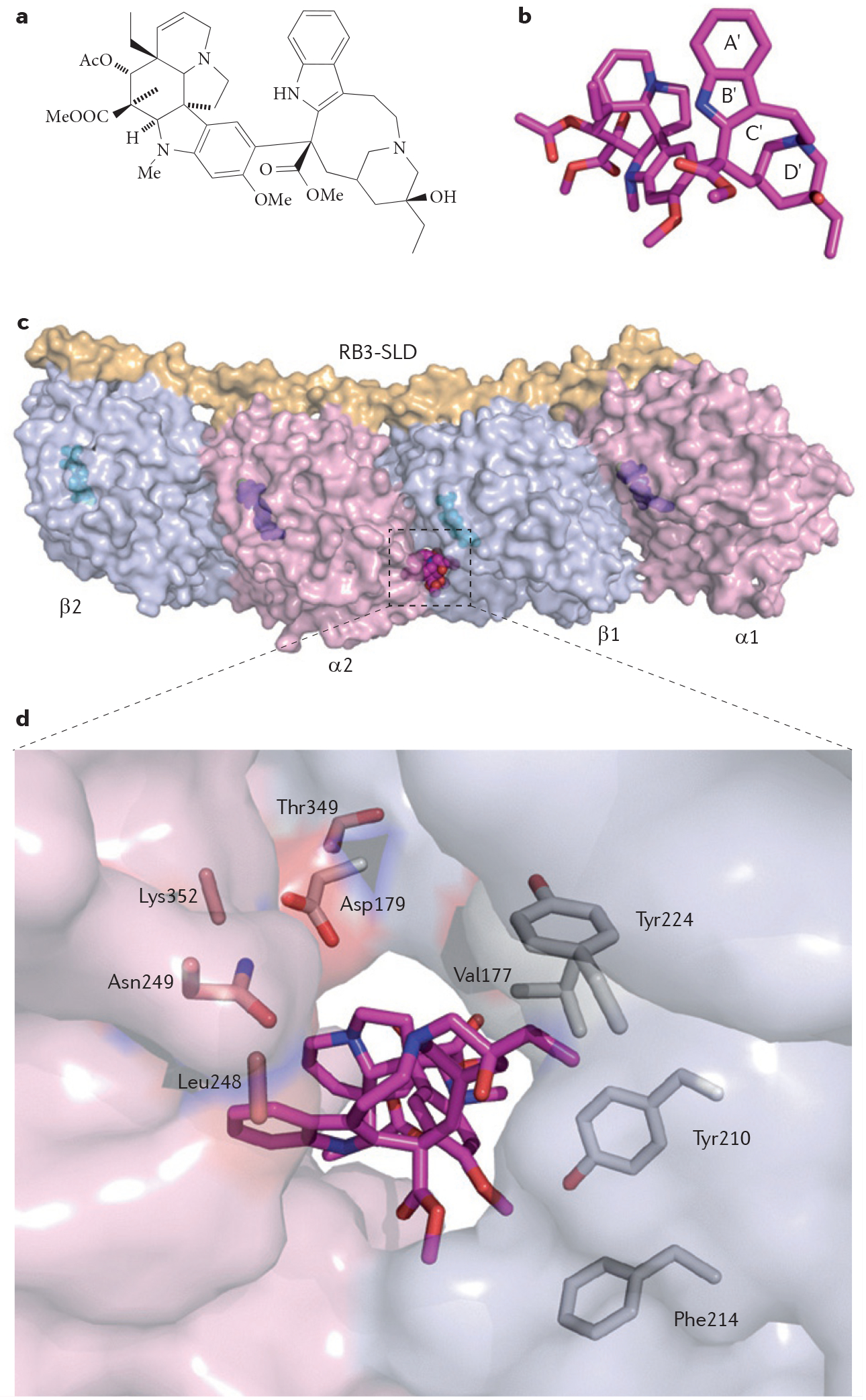
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
