

Efficient mapping and cloning of mutations in zebrafish by low-coverage whole-genome sequencing
- PMID: 22174069
- PMCID: PMC3296239
- DOI: 10.1534/genetics.111.136069
Efficient mapping and cloning of mutations in zebrafish by low-coverage whole-genome sequencing
Abstract
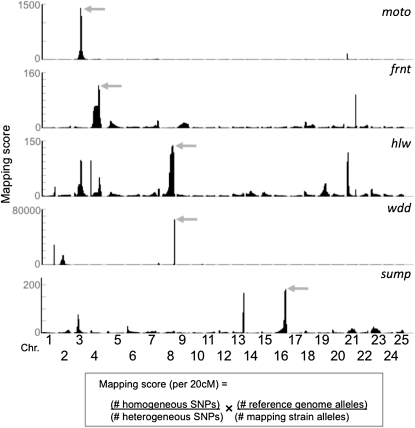
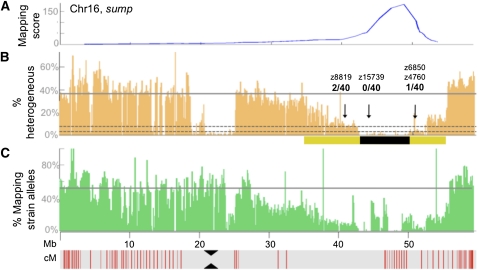
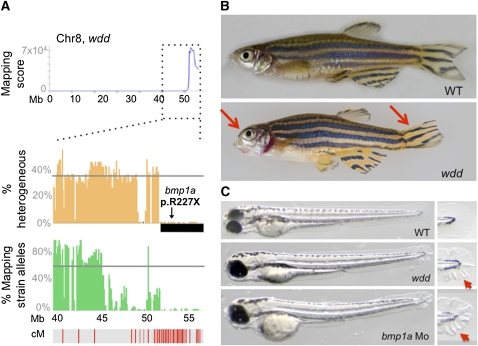
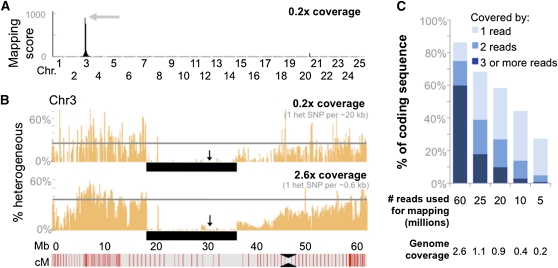
The generation and analysis of mutants in zebrafish has been instrumental in defining the genetic regulation of vertebrate development, physiology, and disease. However, identifying the genetic changes that underlie mutant phenotypes remains a significant bottleneck in the analysis of mutants. Whole-genome sequencing has recently emerged as a fast and efficient approach for identifying mutations in nonvertebrate model organisms. However, this approach has not been applied to zebrafish due to the complicating factors of having a large genome and lack of fully inbred lines. Here we provide a method for efficiently mapping and detecting mutations in zebrafish using these new parallel sequencing technologies. This method utilizes an extensive reference SNP database to define regions of homozygosity-by-descent by low coverage, whole-genome sequencing of pooled DNA from only a limited number of mutant F(2) fish. With this approach we mapped each of the five different zebrafish mutants we sequenced and identified likely causative nonsense mutations in two and candidate mutations in the remainder. Furthermore, we provide evidence that one of the identified mutations, a nonsense mutation in bmp1a, underlies the welded mutant phenotype.
Figures
References
-
- Austin, R. S., D. Vidaurre, G. Stamatiou, R. Breit, N. J. Provart et al, 2011 Next-generation mapping of Arabidopsis genes. Plant J. 67: 715–725. - PubMed
-
- Coe T. S., Hamilton P. B., Griffiths A. M., Hodgson D. J., Wahab M. A., et al. , 2009. Genetic variation in strains of zebrafish (danio rerio) and the implications for ecotoxicology studies. Ecotoxicology 18: 144–150 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
