

Site-specific oxidation of apolipoprotein A-I impairs cholesterol export by ABCA1, a key cardioprotective function of HDL
- PMID: 22178192
- PMCID: PMC3288272
- DOI: 10.1016/j.bbalip.2011.11.011
Site-specific oxidation of apolipoprotein A-I impairs cholesterol export by ABCA1, a key cardioprotective function of HDL
Abstract
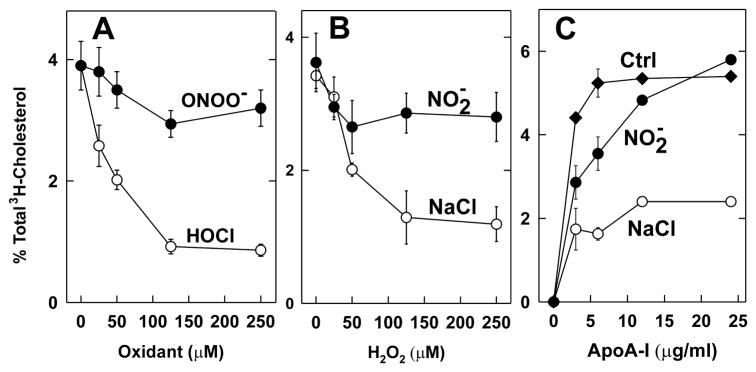
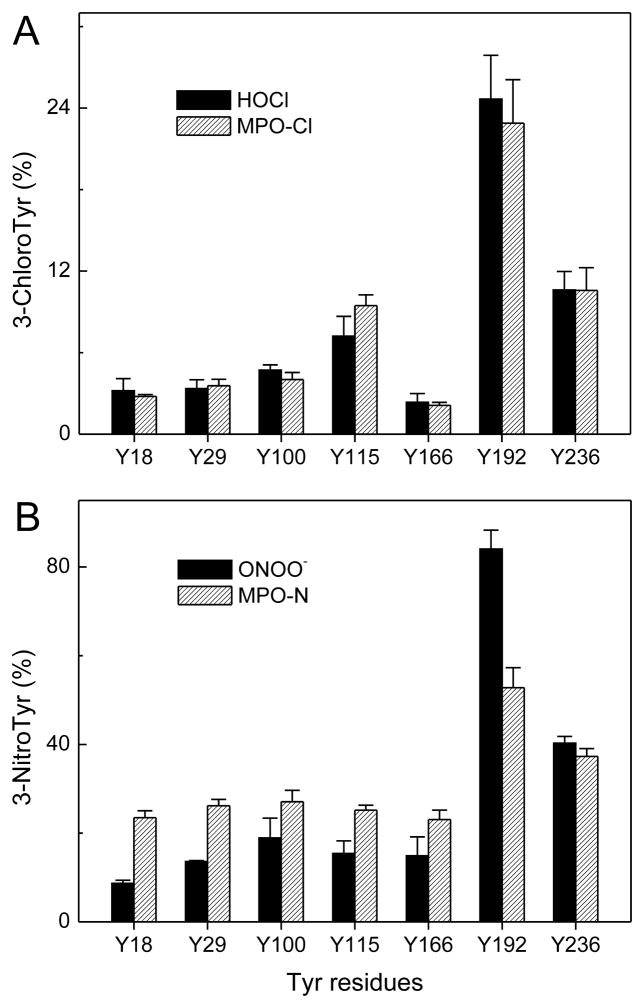
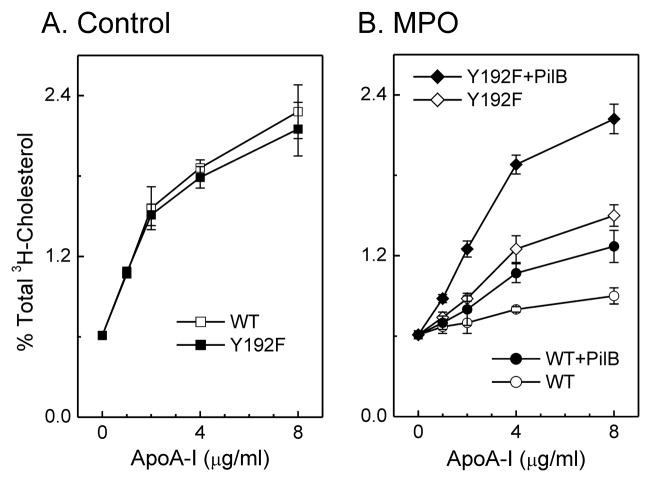
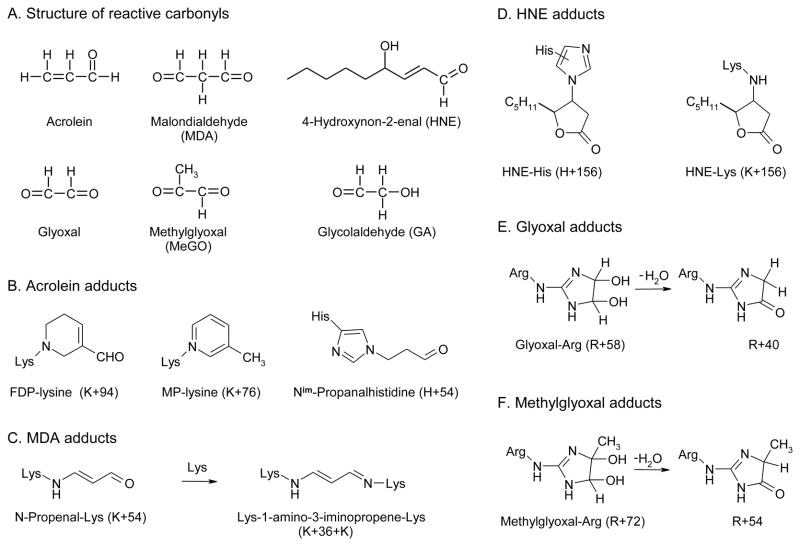
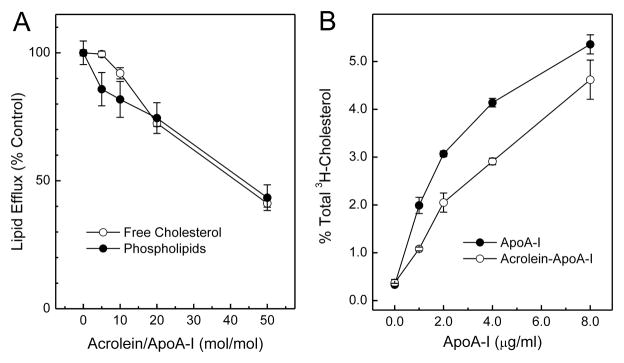
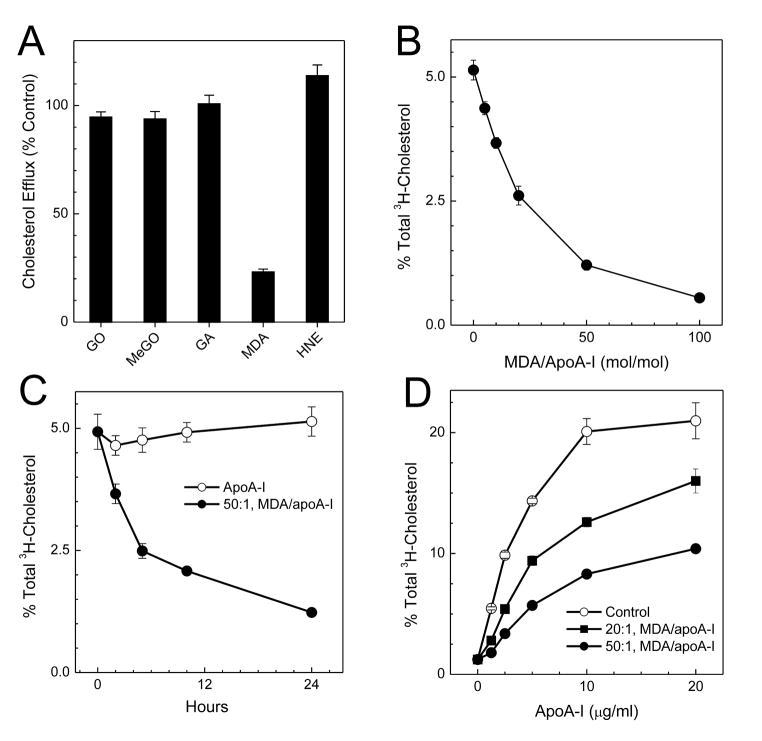
The mechanisms that deprive HDL of its cardioprotective properties are poorly understood. One potential pathway involves oxidative damage of HDL proteins by myeloperoxidase (MPO) a heme enzyme secreted by human artery wall macrophages. Mass spectrometric analysis demonstrated that levels of 3-chlorotyrosine and 3-nitrotyrosine - two characteristic products of MPO - are elevated in HDL isolated from patients with established cardiovascular disease. When apolipoprotein A-I (apoA-I), the major HDL protein, is oxidized by MPO, its ability to promote cellular cholesterol efflux by the membrane-associated ATP-binding cassette transporter A1 (ABCA1) pathway is diminished. Biochemical studies revealed that oxidation of specific tyrosine and methionine residues in apoA-I contributes to this loss of ABCA1 activity. Another potential mechanism for generating dysfunctional HDL involves covalent modification of apoA-I by reactive carbonyls, which have been implicated in atherogenesis and diabetic vascular disease. Indeed, modification of apoA-I by malondialdehyde (MDA) or acrolein also markedly impaired the lipoprotein's ability to promote cellular cholesterol efflux by the ABCA1 pathway. Tandem mass spectrometric analyses revealed that these reactive carbonyls target specific Lys residues in the C-terminus of apoA-I. Importantly, immunochemical analyses showed that levels of MDA-protein adducts are elevated in HDL isolated from human atherosclerotic lesions. Also, apoA-I co-localized with acrolein adducts in such lesions. Thus, lipid peroxidation products might specifically modify HDL in vivo. Our observations support the hypotheses that MPO and reactive carbonyls might generate dysfunctional HDL in humans. This article is part of a Special Issue entitled Advances in High Density Lipoprotein Formation and Metabolism: A Tribute to John F. Oram (1945-2010).
Copyright © 2011 Elsevier B.V. All rights reserved.
Figures
References
-
- Braunwald E. Biomarkers in heart failure. N Engl J Med. 2008;358:2148–2159. - PubMed
-
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. - PubMed
-
- Berliner JA, Heinecke JW. The role of oxidized lipoproteins in atherogenesis. Free Radic Biol Med. 1996;20:707–727. - PubMed
-
- Heinecke JW. Oxidants and antioxidants in the pathogenesis of atherosclerosis: implications for the oxidized low density lipoprotein hypothesis. Atherosclerosis. 1998;141:1–15. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
