

Inhibiting the nucleation of amyloid structure in a huntingtin fragment by targeting α-helix-rich oligomeric intermediates
- PMID: 22178478
- PMCID: PMC3267848
- DOI: 10.1016/j.jmb.2011.12.011
Inhibiting the nucleation of amyloid structure in a huntingtin fragment by targeting α-helix-rich oligomeric intermediates
Abstract
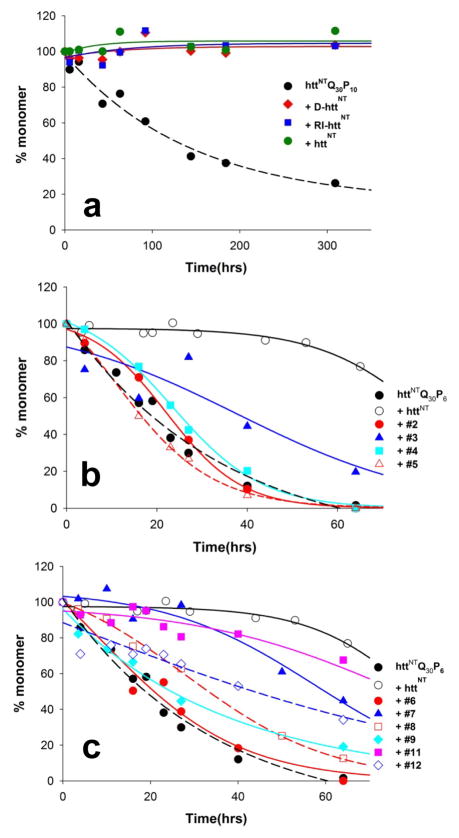
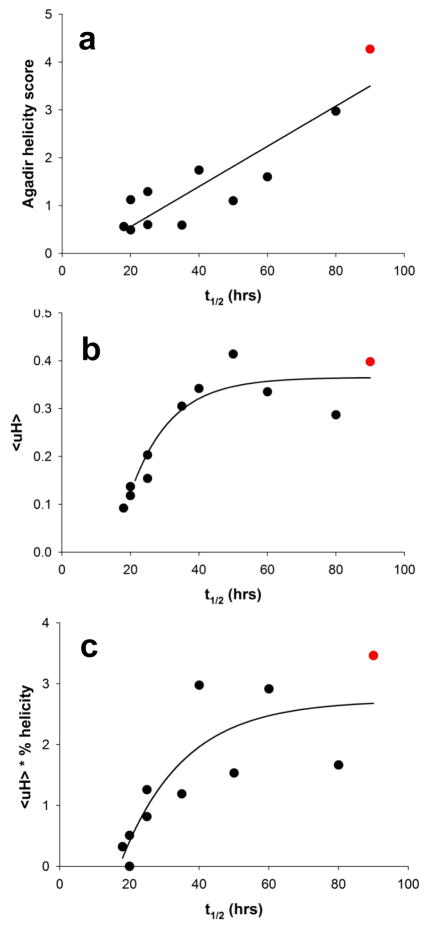
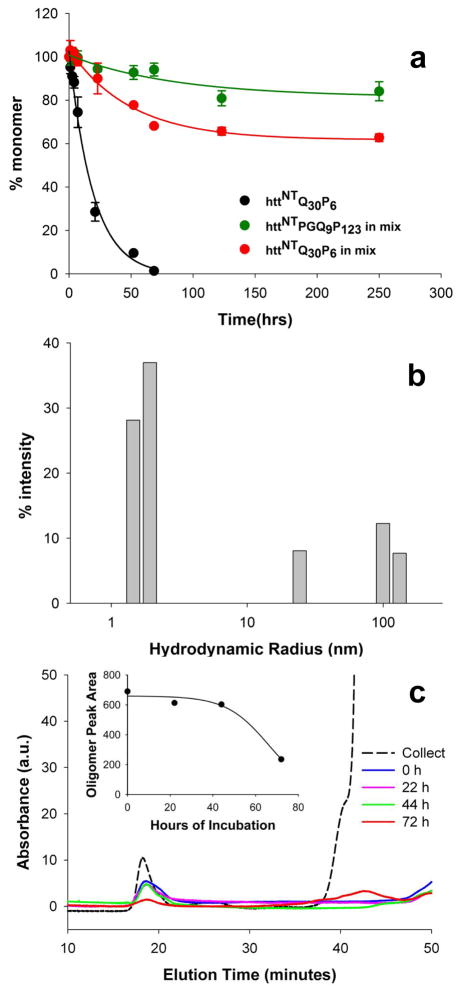
Although oligomeric intermediates are transiently formed in almost all known amyloid assembly reactions, their mechanistic roles are poorly understood. Recently, we demonstrated a critical role for the 17-amino-acid N-terminus (htt(NT) segment) of huntingtin (htt) in the oligomer-mediated amyloid assembly of htt N-terminal fragments. In this mechanism, the htt(NT) segment forms the α-helix-rich core of the oligomers, leaving much of the polyglutamine (polyQ) segment disordered and solvent-exposed. Nucleation of amyloid structure occurs within this local high concentration of disordered polyQ. Here we demonstrate the kinetic importance of htt(NT) self-assembly by describing inhibitory htt(NT)-containing peptides that appear to work by targeting nucleation within the oligomer fraction. These molecules inhibit amyloid nucleation by forming mixed oligomers with the htt(NT) domains of polyQ-containing htt N-terminal fragments. In one class of inhibitors, nucleation is passively suppressed due to the reduced local concentration of polyQ within the mixed oligomer. In the other class, nucleation is actively suppressed by a proline-rich polyQ segment covalently attached to htt(NT). Studies with D-amino acid and scrambled sequence versions of htt(NT) suggest that inhibition activity is strongly linked to the propensity of inhibitory peptides to make amphipathic α-helices. Htt(NT) derivatives with C-terminal cell-penetrating peptide segments also exhibit excellent inhibitory activity. The htt(NT)-based peptides described here, especially those with protease-resistant d-amino acids and/or with cell-penetrating sequences, may prove useful as lead therapeutics for inhibiting the nucleation of amyloid formation in Huntington's disease.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
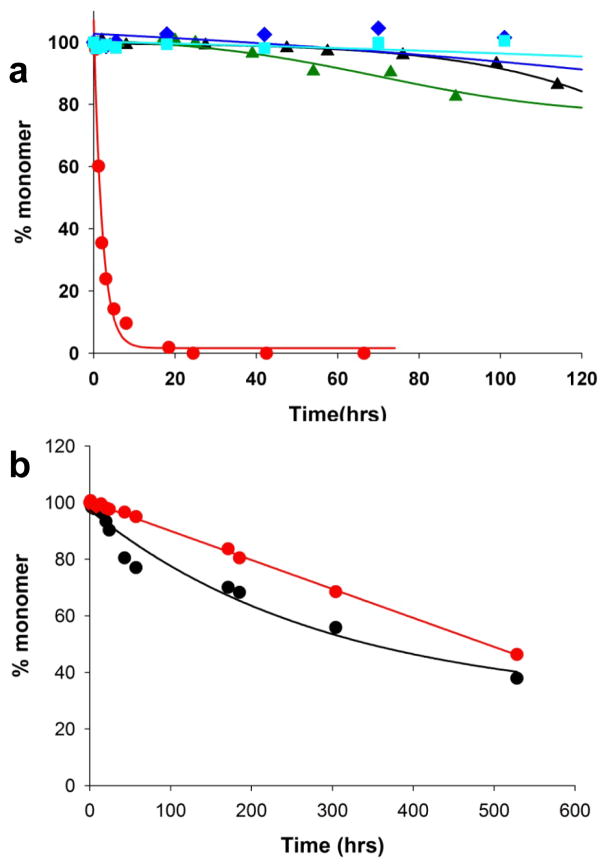
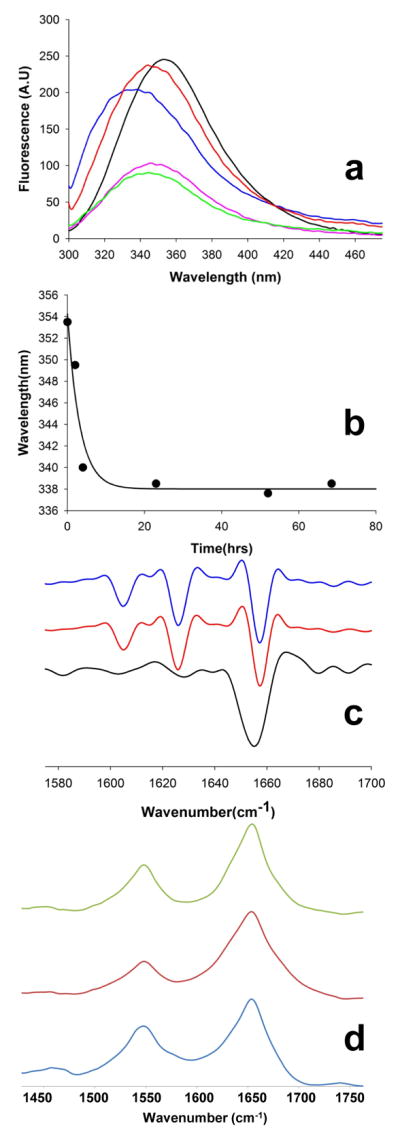
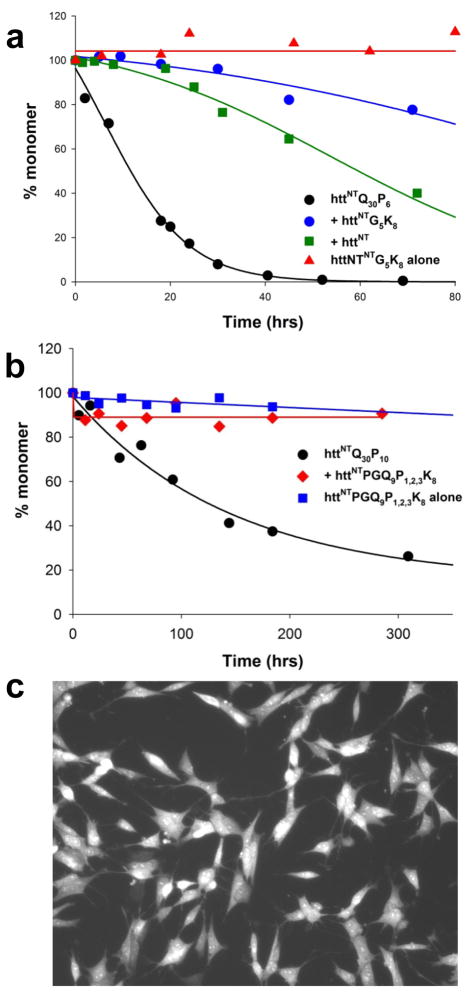
 ); 25.2 μM SWQ37P10K2 (
); 25.2 μM SWQ37P10K2 (
 ); 25.6 μM K2Q35P10K2 (▴); 7.5 μM K2Q41K2 alone (
); 25.6 μM K2Q35P10K2 (▴); 7.5 μM K2Q41K2 alone (
 ); and 7.0 μM K2Q41K2 plus 17.4 μM httNT (
); and 7.0 μM K2Q41K2 plus 17.4 μM httNT (
 ). (b) Aggregation of 18.8 μM of the peptide ESLKSF-Q35-PPPSKETAAAKFERQHMDS incubated alone (●) or with 29.6 μM httNT (
).
). (b) Aggregation of 18.8 μM of the peptide ESLKSF-Q35-PPPSKETAAAKFERQHMDS incubated alone (●) or with 29.6 μM httNT (
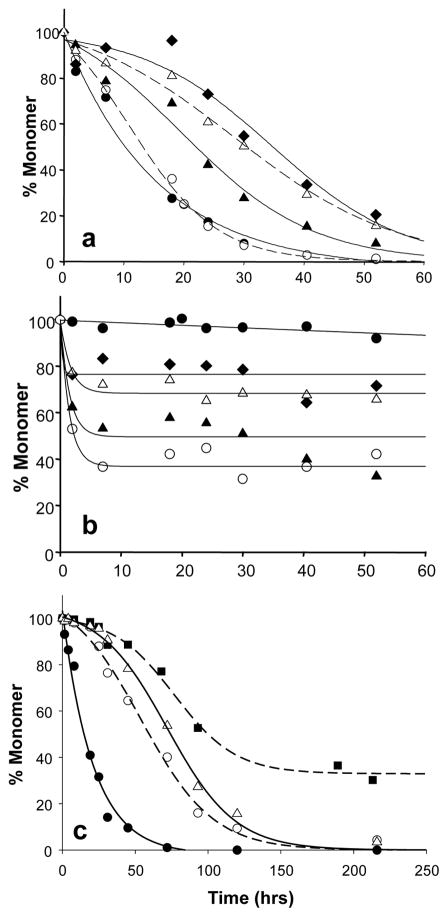
).
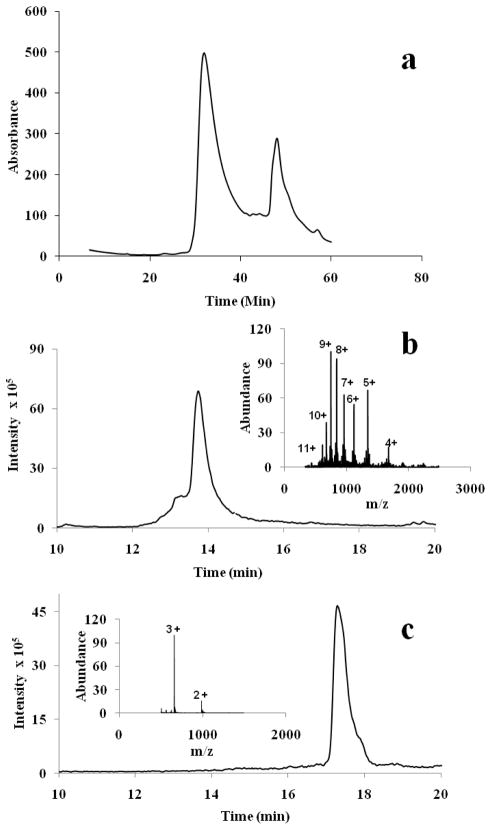
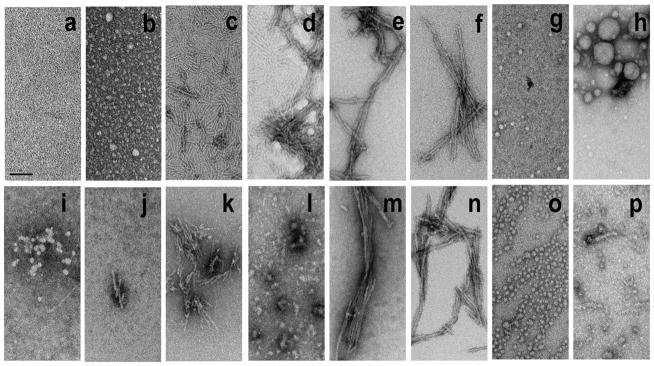
References
-
- Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev. 2010;90:905–81. - PubMed
-
- Wilburn B, Rudnicki DD, Zhao J, Weitz TM, Cheng Y, Gu XF, Greiner E, Park CS, Wang N, Sopher BL, La Spada AR, Osmand A, Margolis RL, Sun YE, Yang XW. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington’s disease-like 2 mice. Neuron. 2011;70:427–440. - PMC - PubMed
-
- Bates GP, Benn C. The polyglutamine diseases. In: Bates GP, Harper PS, Jones L, editors. Huntington’s Disease. Oxford University Press; Oxford, U.K: 2002. pp. 429–472.
-
- Sathasivam K, Lane A, Legleiter J, Warley A, Woodman B, Finkbeiner S, Paganetti P, Muchowski PJ, Wilson S, Bates GP. Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington’s disease. Human Molecular Genetics. 2010;19:65–78. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
