

Performance comparison of whole-genome sequencing platforms
- PMID: 22178993
- PMCID: PMC4076012
- DOI: 10.1038/nbt.2065
Performance comparison of whole-genome sequencing platforms
Erratum in
- Nat Biotechnol. 2012 Jun;30(6):562
Abstract
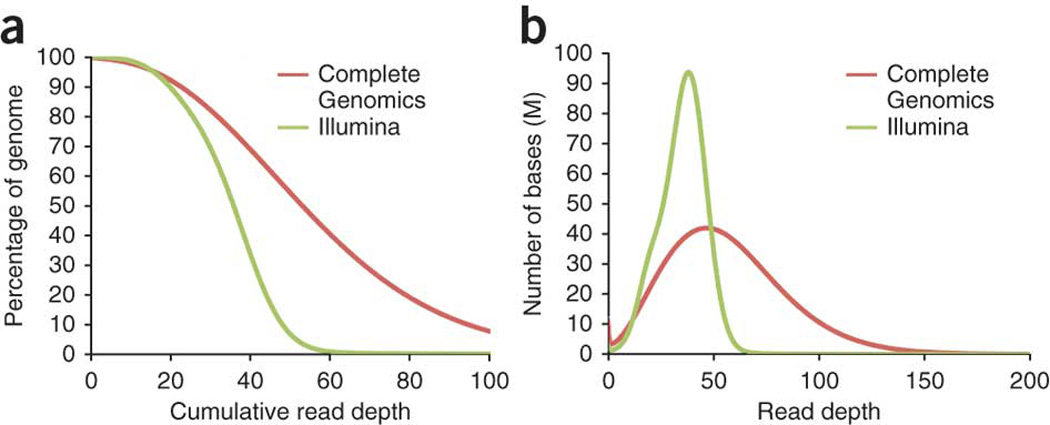
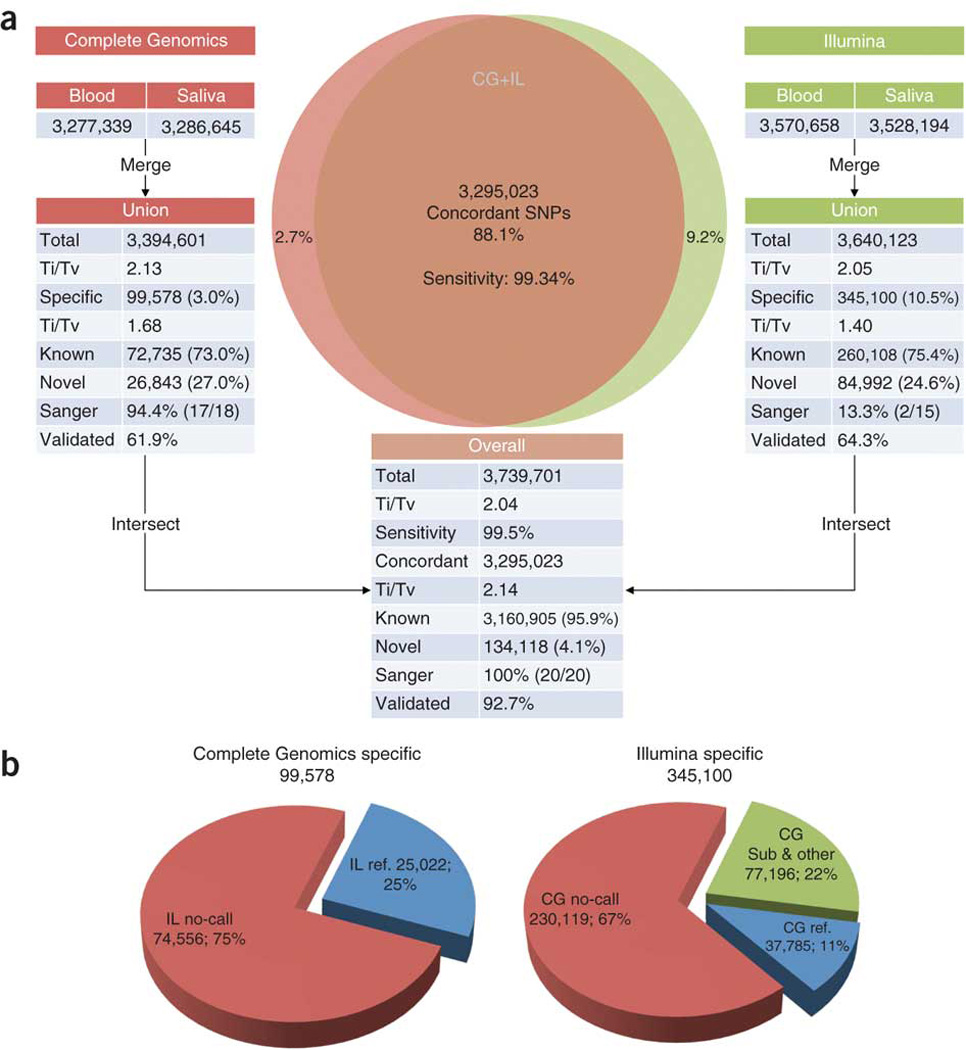
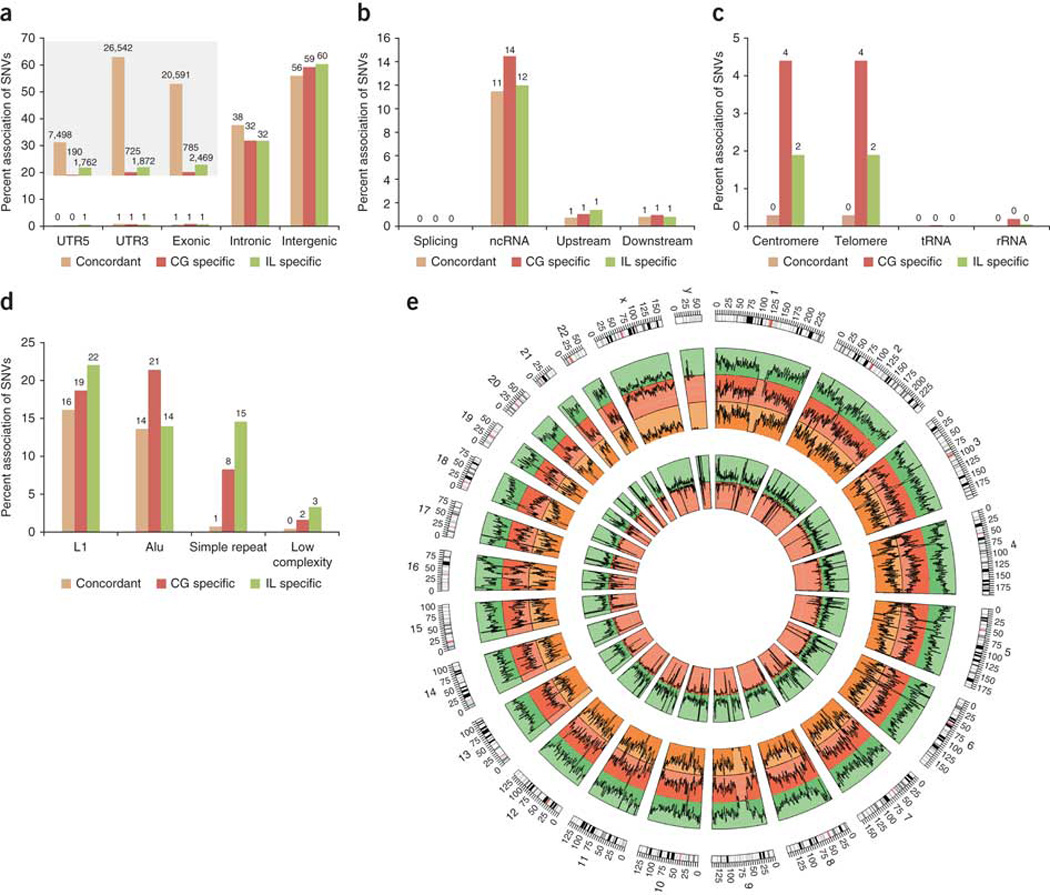
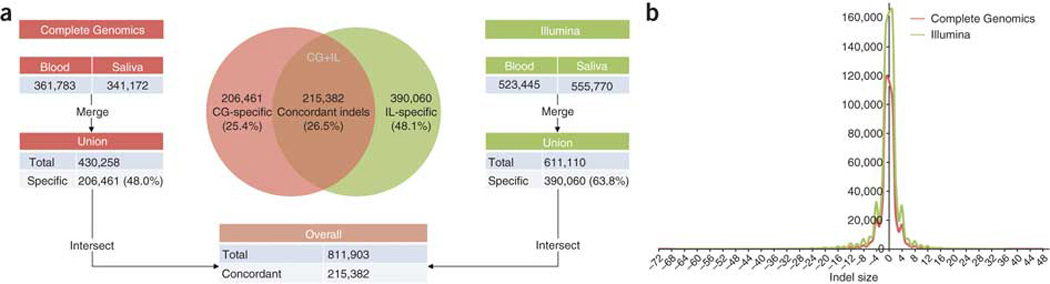
Whole-genome sequencing is becoming commonplace, but the accuracy and completeness of variant calling by the most widely used platforms from Illumina and Complete Genomics have not been reported. Here we sequenced the genome of an individual with both technologies to a high average coverage of ∼76×, and compared their performance with respect to sequence coverage and calling of single-nucleotide variants (SNVs), insertions and deletions (indels). Although 88.1% of the ∼3.7 million unique SNVs were concordant between platforms, there were tens of thousands of platform-specific calls located in genes and other genomic regions. In contrast, 26.5% of indels were concordant between platforms. Target enrichment validated 92.7% of the concordant SNVs, whereas validation by genotyping array revealed a sensitivity of 99.3%. The validation experiments also suggested that >60% of the platform-specific variants were indeed present in the genome. Our results have important implications for understanding the accuracy and completeness of the genome sequencing platforms.
Figures
References
-
- Wheeler DA, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
