

The adaptor protein CRK is a pro-apoptotic transducer of endoplasmic reticulum stress
- PMID: 22179045
- PMCID: PMC3245775
- DOI: 10.1038/ncb2395
The adaptor protein CRK is a pro-apoptotic transducer of endoplasmic reticulum stress
Abstract
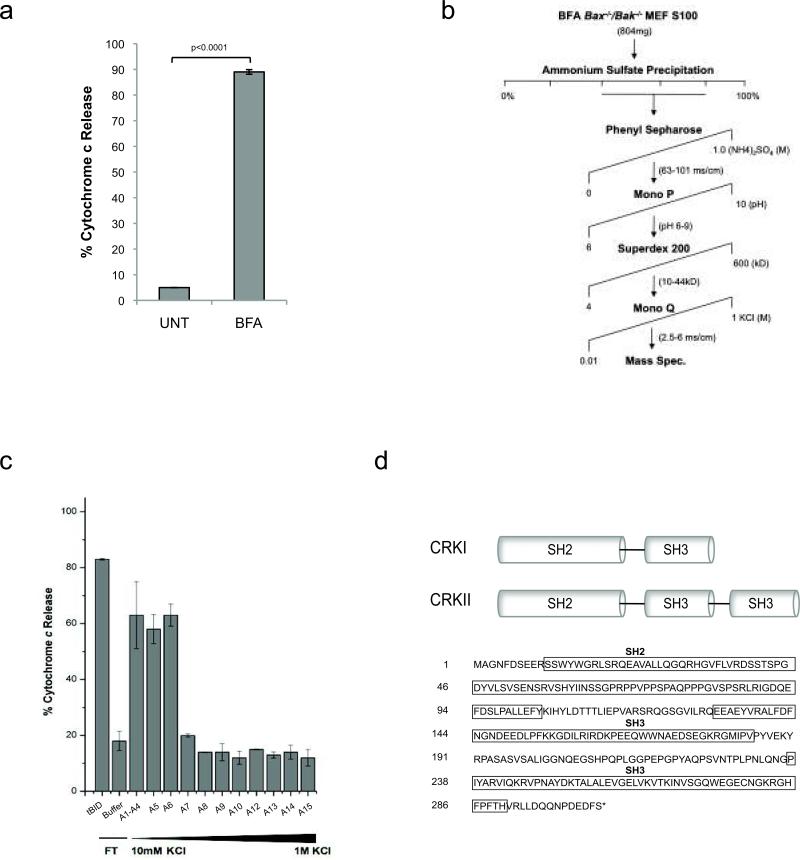
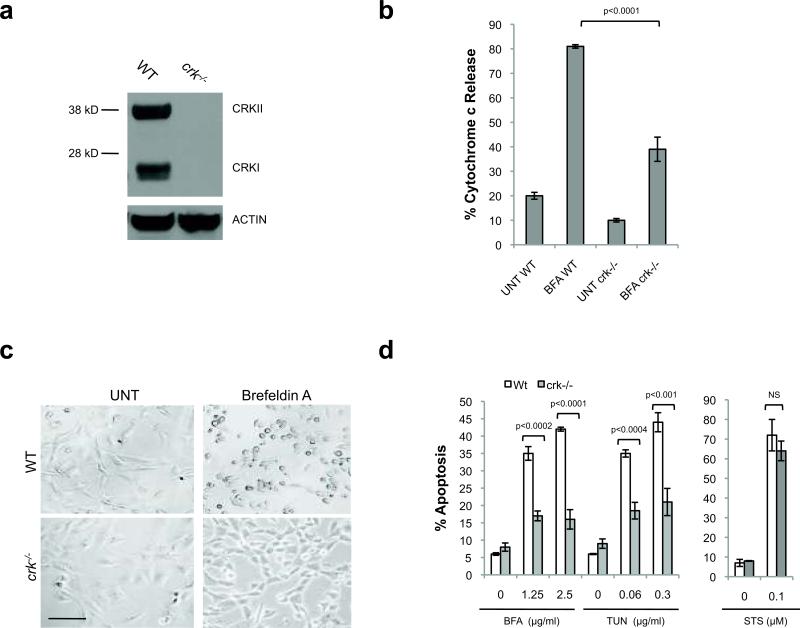
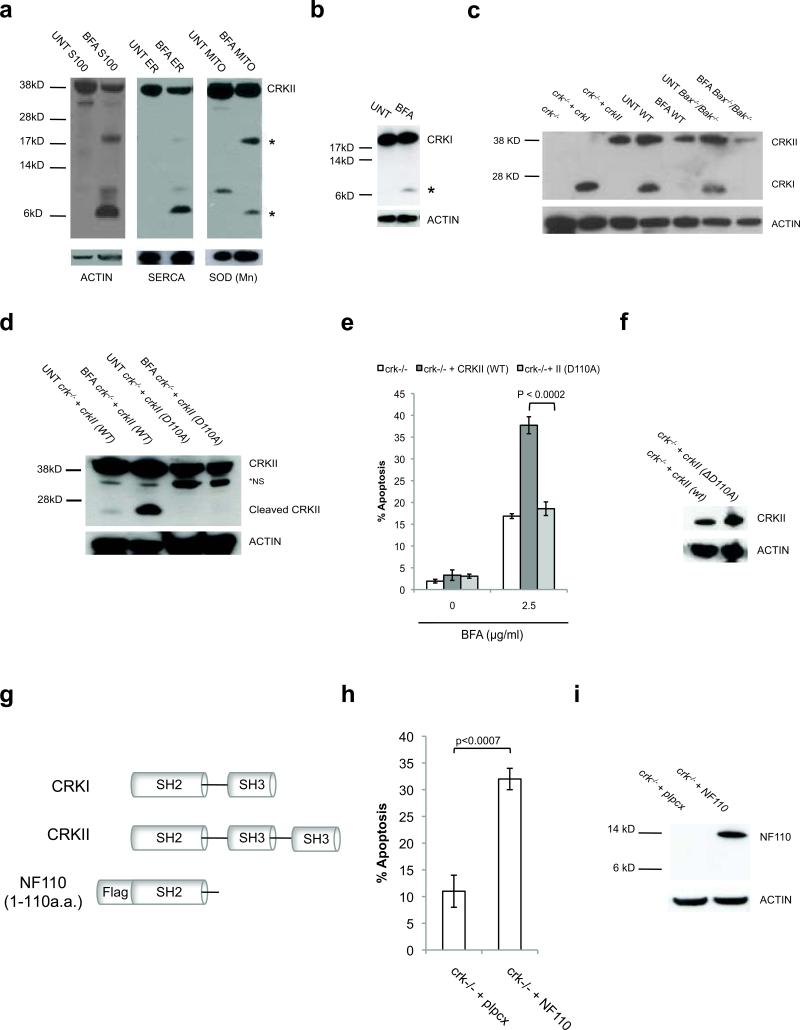
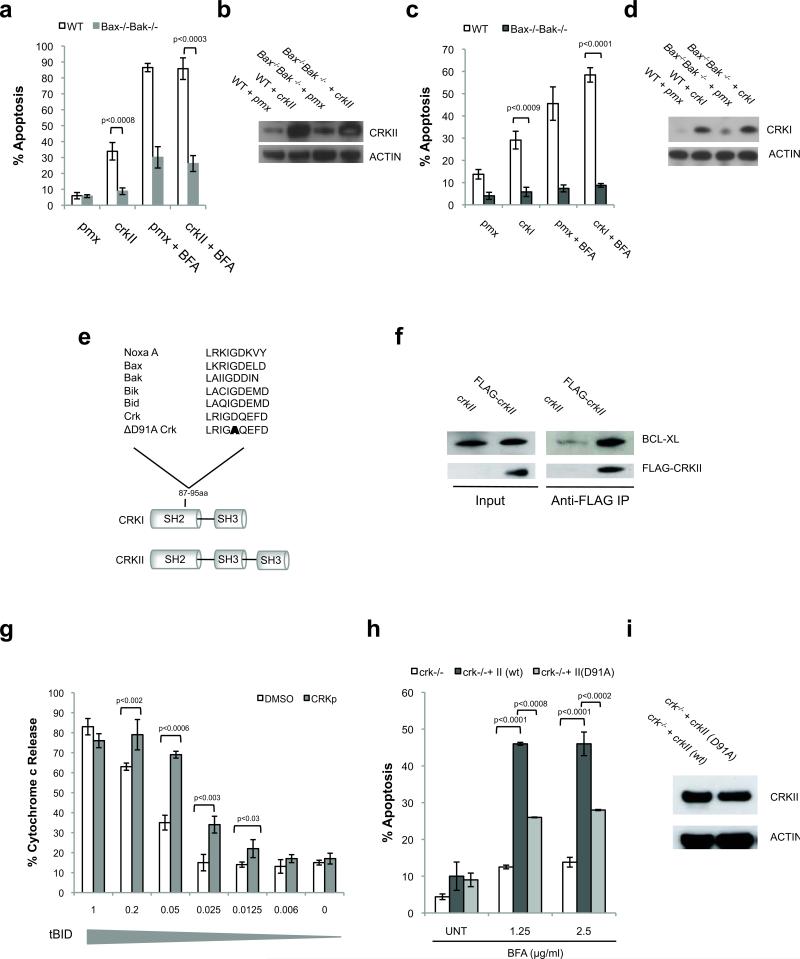
Excessive demands on the protein-folding capacity of the endoplasmic reticulum (ER) cause irremediable ER stress and contribute to cell loss in a number of cell degenerative diseases, including type 2 diabetes and neurodegeneration. The signals communicating catastrophic ER damage to the mitochondrial apoptotic machinery remain poorly understood. We used a biochemical approach to purify a cytosolic activity induced by ER stress that causes release of cytochrome c from isolated mitochondria. We discovered that the principal component of the purified pro-apoptotic activity is the proto-oncoprotein CRK (CT10-regulated kinase), an adaptor protein with no known catalytic activity. Crk(-/-) cells are strongly resistant to ER-stress-induced apoptosis. Moreover, CRK is cleaved in response to ER stress to generate an amino-terminal M(r)~14K fragment with greatly enhanced cytotoxic potential. We identified a putative BH3 (BCL2 homology 3) domain within this N-terminal CRK fragment, which sensitizes isolated mitochondria to cytochrome c release and when mutated significantly reduces the apoptotic activity of CRK in vivo. Together these results identify CRK as a pro-apoptotic protein that signals irremediable ER stress to the mitochondrial execution machinery.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
