

doi: 10.1038/nmeth.1810.
Detection of structural variants and indels within exome data
Affiliations
- PMID: 22179552
- PMCID: PMC3269549
- DOI: 10.1038/nmeth.1810
Item in Clipboard
Detection of structural variants and indels within exome data
Nat Methods.
.
Abstract
We report an algorithm to detect structural variation and indels from 1 base pair (bp) to 1 Mbp within exome sequence data sets. Splitread uses one end-anchored placements to cluster the mappings of subsequences of unanchored ends to identify the size, content and location of variants with high specificity and sensitivity. The algorithm discovers indels, structural variants, de novo events and copy number-polymorphic processed pseudogenes missed by other methods.
Conflict of interest statement
E.E.E. is a member of the Scientific Advisory Board of Pacific Biosciences.
Figures
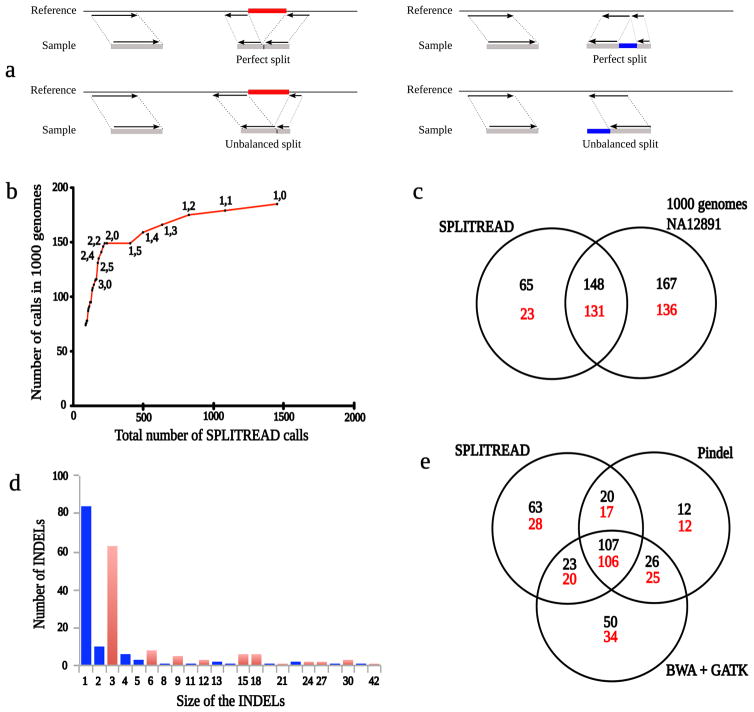
(A) Schematic diagrams for the mapping of paired-end sequences in cases where an individual has either a deletion (red) or an insertion (blue) with respect to the reference sequence. In each case, one-end anchored sequence is used to map one read in a pair. The second (unmapped) read is then decomposed into either two equal subsequences (balanced split) or two unequal subsequences (unbalanced split). (B) Number of Splitread predictions called by 1000 Genomes plotted against the total number of Splitread predictions using the indicated threshold numbers of balanced and unbalanced reads, respectively. A threshold of two balanced and two unbalanced splits maximizes intersection with 1000 Genomes Project calls without losing any positive predictive value. (C) A Venn diagram comparing variants detected by Splitread exome analysis versus whole-genome sequence analysis of NA12891 (black) or all variants within dbSNP130 (red). In order to intersect, variants must be at the same position and within 10 base pairs of the predicted size. (D) Length distribution of insertions and deletions mapping within the coding region of NA12891 as predicted by Splitread. Events with multiples of three base pairs (red) are compared to those that would disrupt the frame (blue). (E) A Venn diagram comparing Pindel, GATK and Splitread call sets on NA12891. The total number of events (black) is compared to those previously detected (red) as part of dbSNP130 and/or the 1000 Genomes Project.
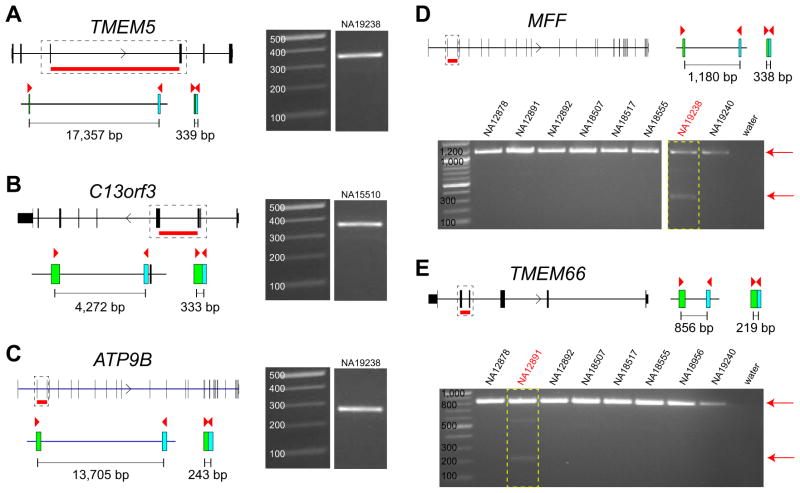
Gene models and predicted intron deletions of the processed pseudogenes are shown. Primers (red triangles) are designed in the coding region of the genes and the expected product size for the processed pseudogenes are shown for (A) TMEM5, (B) C13orf3, (C) ATP9B, (D) MFF, and (E) TMEM66. Gel images show the size of the amplified product. We were able to detect the processed version of these genes in our PCR experiments. In D-E we genotyped the processed pseudogenes MFF and TMEM66 within eight HapMap samples and show that each is amplified only in the predicted sample [boxed in red: NA19238 (MFF) and NA12891 (TMEM66)]. All PCRs amplify the normal gene (signal on the top) with only one sample each amplifying the processed gene.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
