

Joining forces: integrating proteomics and cross-linking with the mass spectrometry of intact complexes
- PMID: 22180098
- PMCID: PMC3316738
- DOI: 10.1074/mcp.R111.014027
Joining forces: integrating proteomics and cross-linking with the mass spectrometry of intact complexes
Abstract
Protein assemblies are critical for cellular function and understanding their physical organization is the key aim of structural biology. However, applying conventional structural biology approaches is challenging for transient, dynamic, or polydisperse assemblies. There is therefore a growing demand for hybrid technologies that are able to complement classical structural biology methods and thereby broaden our arsenal for the study of these important complexes. Exciting new developments in the field of mass spectrometry and proteomics have added a new dimension to the study of protein-protein interactions and protein complex architecture. In this review, we focus on how complementary mass spectrometry-based techniques can greatly facilitate structural understanding of protein assemblies.
Figures


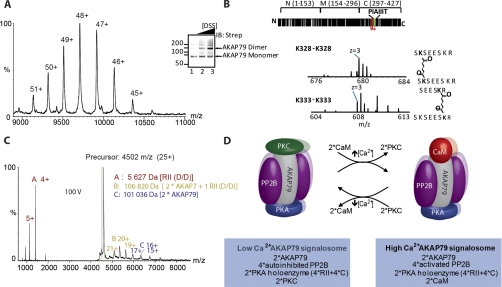
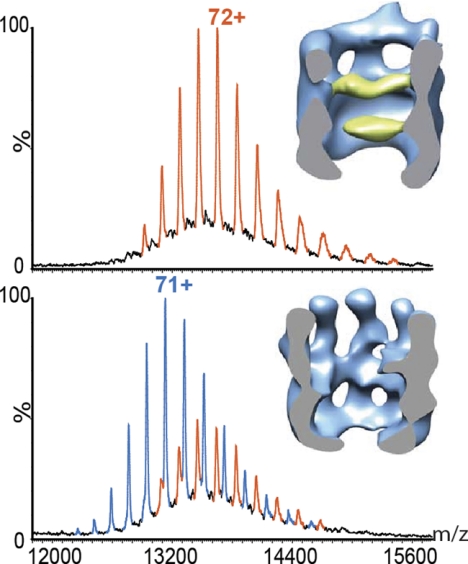

References
-
- Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 - PubMed
-
- Gstaiger M., Aebersold R. (2009) Applying mass spectrometry-based proteomics to genetics, genomics and network biology. Nat. Rev. 10, 617–627 - PubMed
-
- Cox J., Mann M. (2011) Quantitative, high-resolution proteomics for data-driven systems biology. Annu. Rev. Biochem. 80, 273–299 - PubMed
-
- Gingras A. C., Gstaiger M., Raught B., Aebersold R. (2007) Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 8, 645–654 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
