

Checkpoint kinase 1 (Chk1)-short is a splice variant and endogenous inhibitor of Chk1 that regulates cell cycle and DNA damage checkpoints
- PMID: 22184239
- PMCID: PMC3252905
- DOI: 10.1073/pnas.1104767109
Checkpoint kinase 1 (Chk1)-short is a splice variant and endogenous inhibitor of Chk1 that regulates cell cycle and DNA damage checkpoints
Abstract
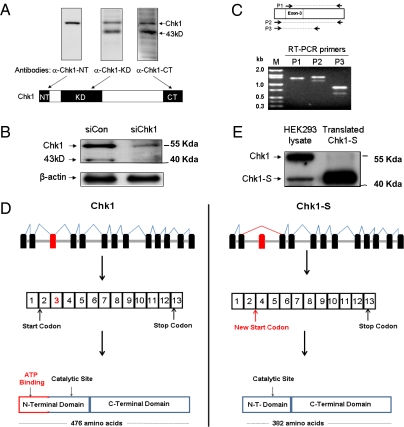
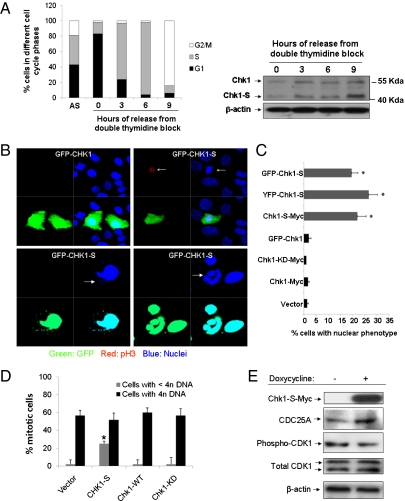
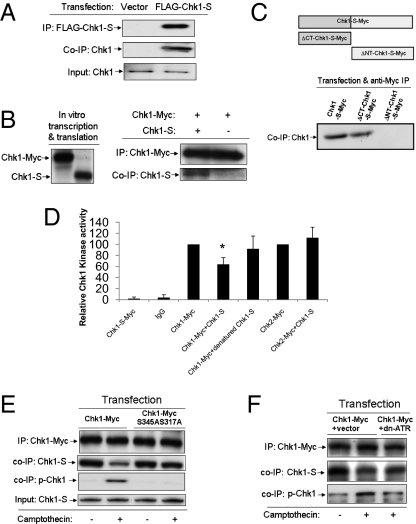
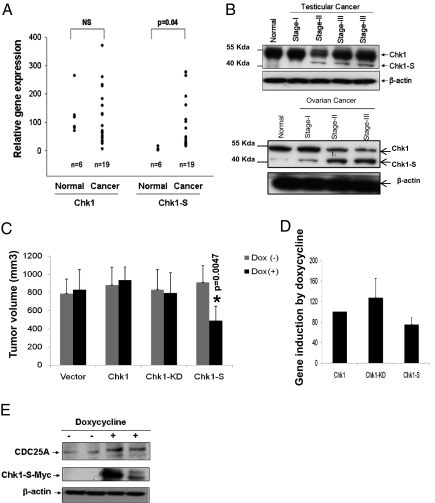
Checkpoint kinase 1 (Chk1) is a key regulator of checkpoint signaling in both the unperturbed cell cycle and DNA damage response. Under these conditions, Chk1 becomes active to prevent premature CDK1 activation and mitotic entry until DNA is properly replicated or repaired. It is unclear how Chk1 activity is controlled in the unperturbed cell cycle. During DNA damage, Chk1 is activated by ataxia telangiectasia and Rad3 related (ATR)-mediated phosphorylation; however, it is not entirely clear how this phosphorylation results in Chk1 activation. Here we report an N-terminally truncated alternative splice variant of Chk1, Chk1-S. Importantly, we show that Chk1-S is an endogenous repressor and regulator of Chk1. In the unperturbed cell cycle, Chk1-S interacts with and antagonizes Chk1 to promote the S-to-G2/M phase transition. During DNA damage, Chk1 is phosphorylated, which disrupts the Chk1-Chk1-S interaction, resulting in free, active Chk1 to arrest the cell cycle and facilitate DNA repair. Higher levels of Chk1-S are expressed, along with Chk1, in fetal and cancer tissues than in normal tissues. However, forced overexpression of Chk1-S in cultured cells and tumor xenografts induces premature mitotic entry, mitotic catastrophe, and reduction of tumor growth. The identification of Chk1-S as a unique splice variant and key regulator of Chk1 provides insights into cell cycle regulation and DNA damage response.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
Sibling rivalry in checkpoint control of cell cycle and DNA damage response.Cell Cycle. 2012 May 15;11(10):1866-7. doi: 10.4161/cc.20416. Epub 2012 May 15. Cell Cycle. 2012. PMID: 22544330 No abstract available.
References
-
- Nurse P. A long twentieth century of the cell cycle and beyond. Cell. 2000;100(1):71–78. - PubMed
-
- Paulovich AG, Toczyski DP, Hartwell LH. When checkpoints fail. Cell. 1997;88:315–321. - PubMed
-
- Zhou BB, Elledge SJ. The DNA damage response: Putting checkpoints in perspective. Nature. 2000;408:433–439. - PubMed
-
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. - PubMed
-
- Sanchez Y, et al. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–1501. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
