

Pathological basis of symptoms and crises in sickle cell disorder: implications for counseling and psychotherapy
- PMID: 22184515
- PMCID: PMC3222266
- DOI: 10.4081/hr.2010.e2
Pathological basis of symptoms and crises in sickle cell disorder: implications for counseling and psychotherapy
Abstract
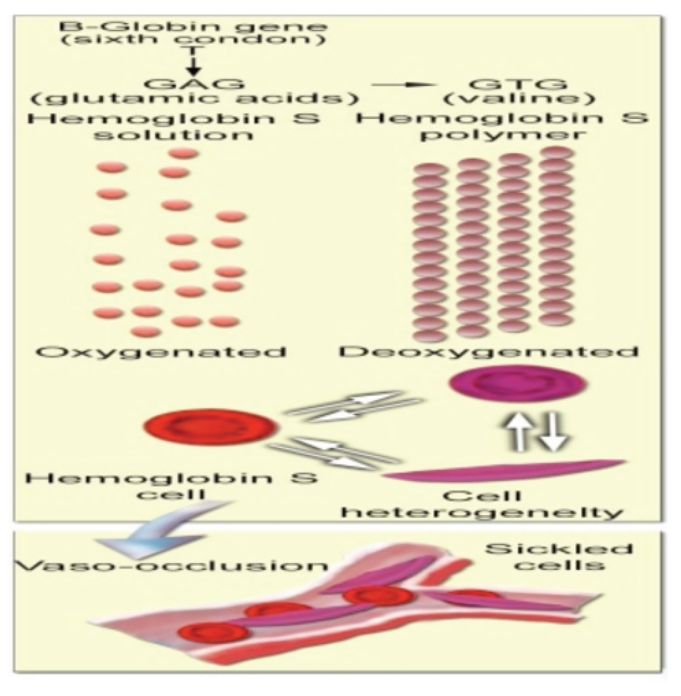
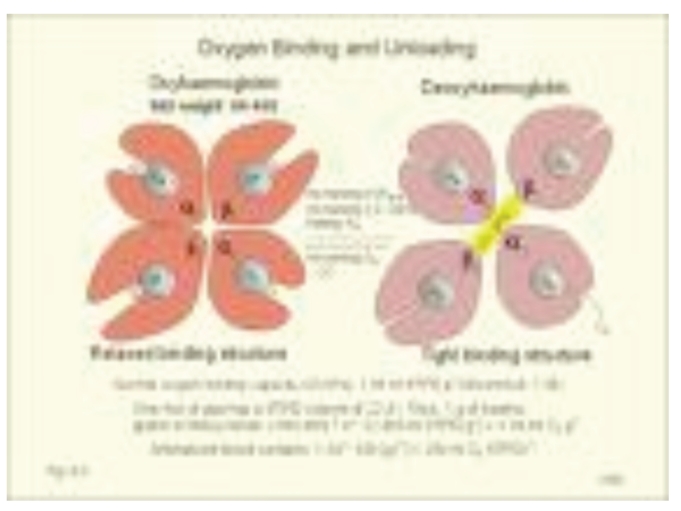
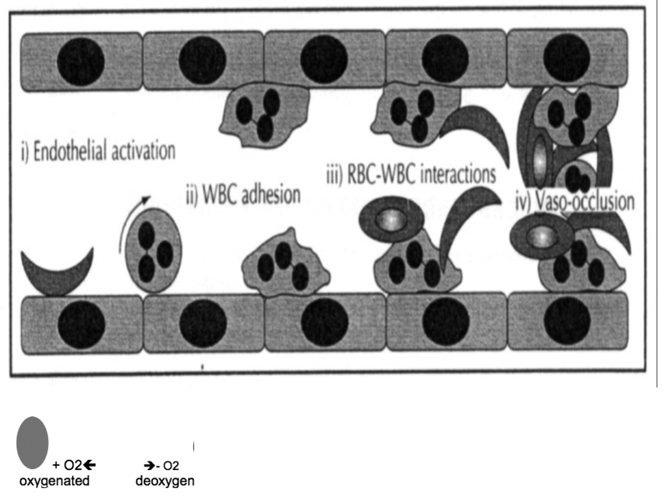
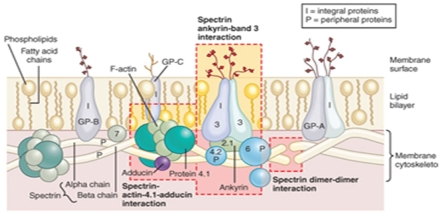
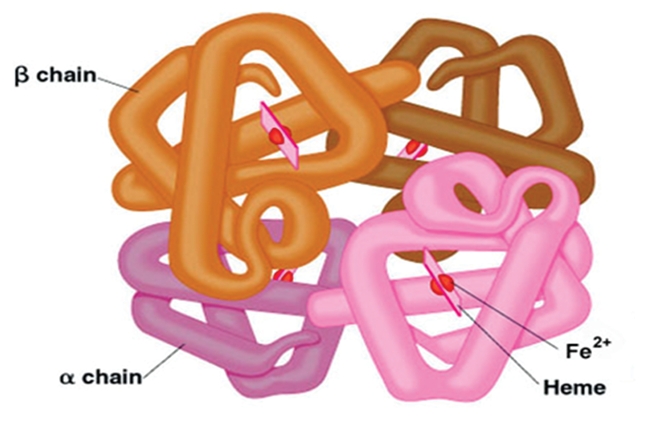
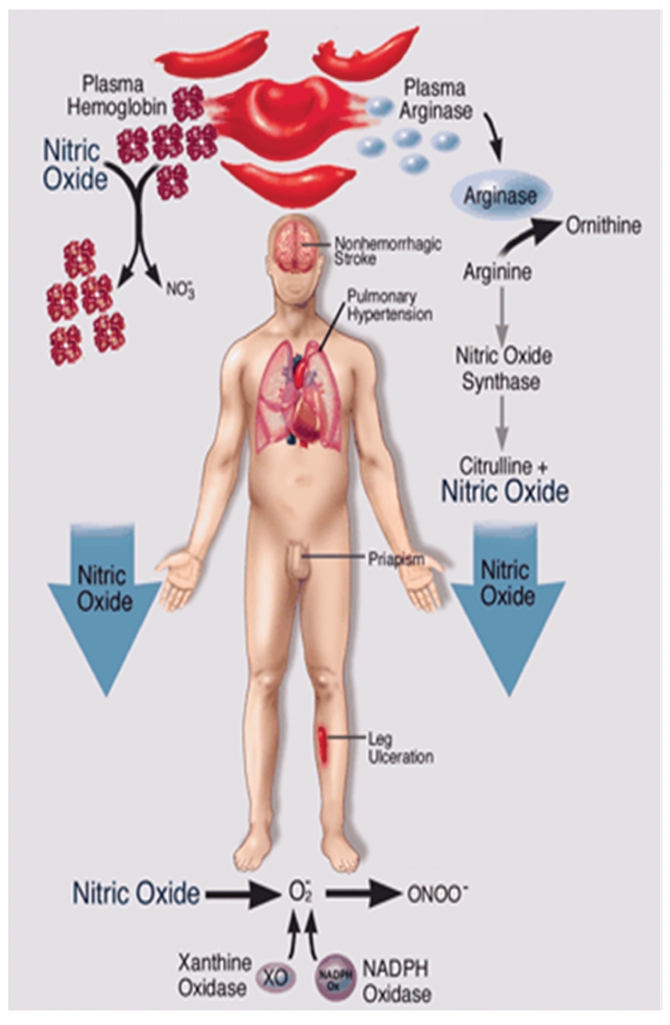
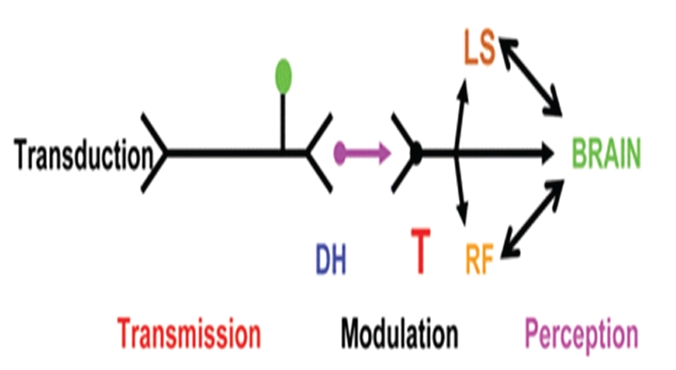
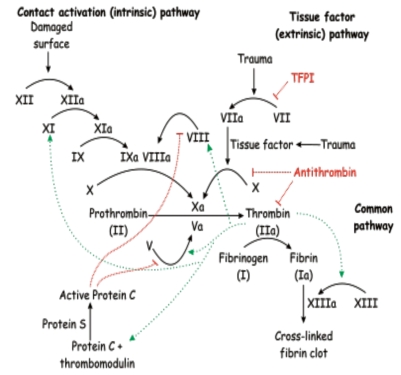
Sickle Cell Disorder (SCD) is a congenital hemoglobinopathy. There is little in literature regarding the psychological variables affecting individuals living with SCD and all of the significant people around them. There are also limited numbers of trained clinical psychologists and genetic counselors to cater for the psychotherapeutic needs of individuals living with SCD. Even among those who have been trained, only a few might have fully grasped the complexities of the disease pathology.Early understanding of its pathological nature, sources, types, complications, pathophysiological basis, and clinical severity of symptoms among clinical psychologists, genetic counselors and psychotherapists, as well as general medical practitioners, could guide them in providing holistic care for dealing with and reducing pain among individuals living with SCD. It could allow risk-based counseling for families and individuals. It could also justify the early use of disease-modifying or curative interventions, such as hydroxyurea (HU), chronic transfusions (CTs), or stem-cell transplantation (SCT) by general medical practitioners. Hence, the need for this paper on the pathophysiology of SCD.
Keywords: counseling and psychotherapy.; implications; pathology; sickle cell disorder.
Figures
References
-
- Embury SH, Hebbel RP, Mohandas N, Steinberg MH, editors. Basic Principles and Clinical Picture. New York: Raven Press; 1994. Sickle cell disease; pp. 311–311.
-
- Serjeant GR. 3rd ed. New York: Oxford University Press; 2001. Sickle cell disease.
-
- Ballas SK. Progress in Pain Research and Management. Vol. 11. Seattle, WA: IASP Press; 1998. Sickle Cell Pain.
-
- Benjamin LJ, Dampier CD, Jacox A, et al. American Pain Society Clinical Practice Guidelines Series No. 1. Glenview, IL: 1999. Guideline for the Management of Acute and Chronic Pain in Sickle cell disease.
-
- Benjamin LJ. Nature and treatment of the acute painful episode in sickle cell disease. In: Steinberg MH, et al., editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Cambridge: 2001. pp. 671–710.
LinkOut - more resources
Full Text Sources
Research Materials
