

Comprehensive analysis of protein modifications by top-down mass spectrometry
- PMID: 22187450
- PMCID: PMC3320739
- DOI: 10.1161/CIRCGENETICS.110.957829
Comprehensive analysis of protein modifications by top-down mass spectrometry
Abstract
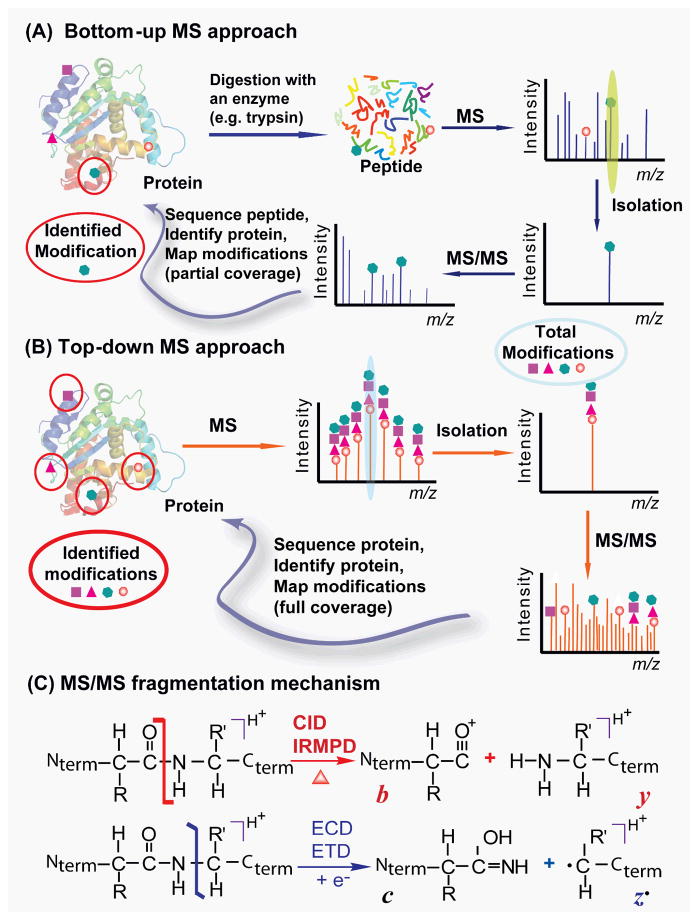
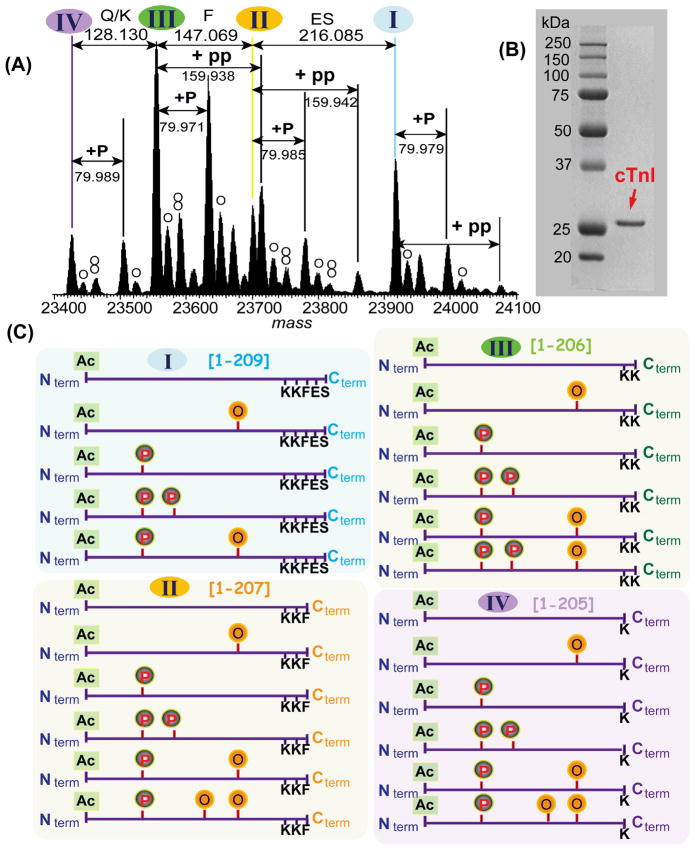
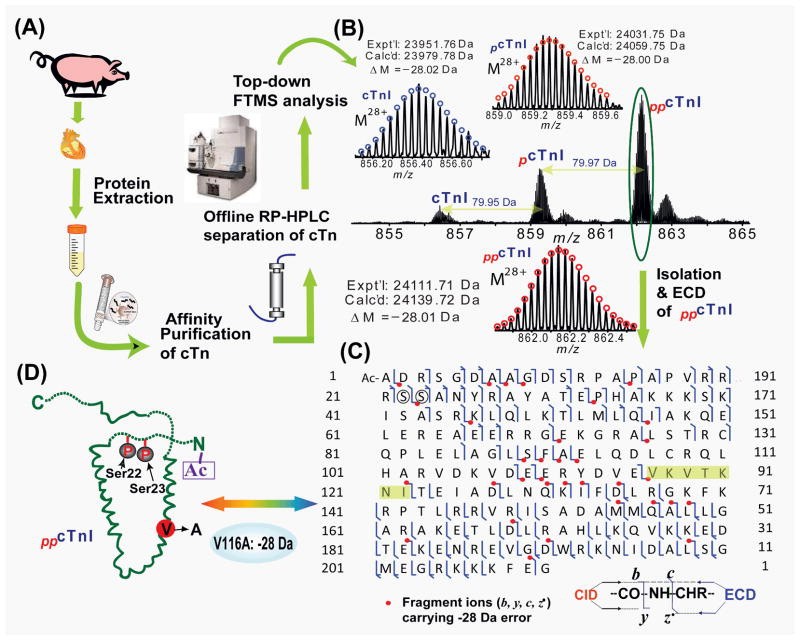
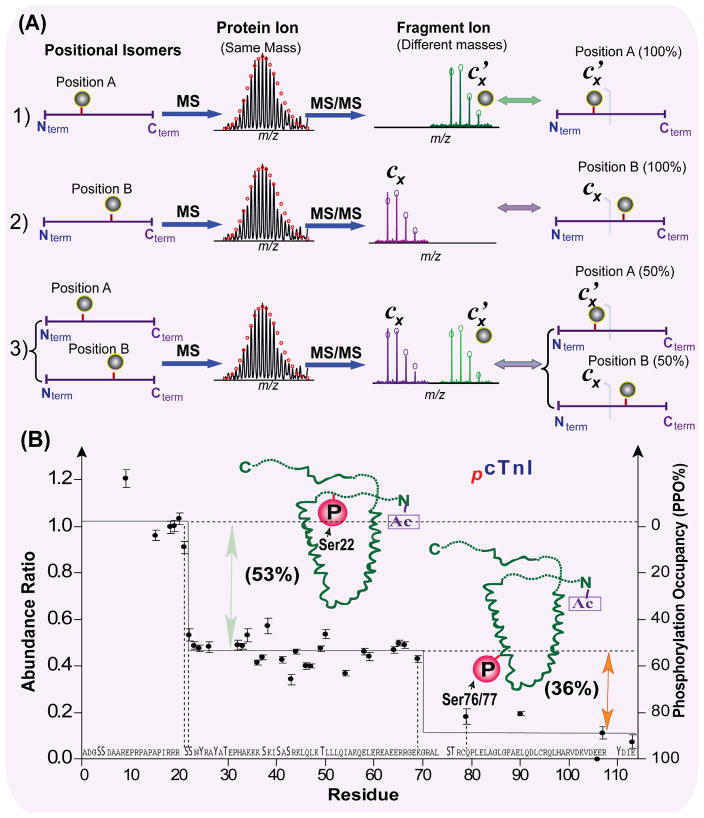
Mass spectrometry (MS)-based proteomics is playing an increasingly important role in cardiovascular research. Proteomics includes identification and quantification of proteins and the characterization of protein modifications, such as posttranslational modifications and sequence variants. The conventional bottom-up approach, involving proteolytic digestion of proteins into small peptides before MS analysis, is routinely used for protein identification and quantification with high throughput and automation. Nevertheless, it has limitations in the analysis of protein modifications, mainly because of the partial sequence coverage and loss of connections among modifications on disparate portions of a protein. An alternative approach, top-down MS, has emerged as a powerful tool for the analysis of protein modifications. The top-down approach analyzes whole proteins directly, providing a "bird's-eye" view of all existing modifications. Subsequently, each modified protein form can be isolated and fragmented in the mass spectrometer to locate the modification site. The incorporation of the nonergodic dissociation methods, such as electron-capture dissociation (ECD), greatly enhances the top-down capabilities. ECD is especially useful for mapping labile posttranslational modifications that are well preserved during the ECD fragmentation process. Top-down MS with ECD has been successfully applied to cardiovascular research, with the unique advantages in unraveling the molecular complexity, quantifying modified protein forms, complete mapping of modifications with full-sequence coverage, discovering unexpected modifications, identifying and quantifying positional isomers, and determining the order of multiple modifications. Nevertheless, top-down MS still needs to overcome some technical challenges to realize its full potential. Herein, we reviewed the advantages and challenges of the top-down method, with a focus on its application in cardiovascular research.
Conflict of interest statement
Dr. Han Zhang and Dr. Ying Ge have no potential conflicts of interest.
Figures
References
-
- Arrell DK, Neverova I, Van Eyk JE. Cardiovascular proteomics - evolution and potential. Circ Res. 2001;88:763–773. - PubMed
-
- Arab S, Gramolini AO, Ping PP, Kislinger T, Stanley B, van Eyk J, Ouzounian M, MacLennan DH, Emili A, Liu PP. Cardiovascular proteomics - tools to develop novel biomarkers and potential applications. J Am Coll Cardiol. 2006;48:1733–1741. - PubMed
-
- White MY, Van Eyk JE. Cardiovascular proteomics - past, present, and future. Mol Diag Ther. 2007;11:83–95. - PubMed
-
- Edwards AVG, White MY, Cordwell SJ. The role of proteomics in clinical cardiovascular biomarker discovery. Mol Cell Proteomics. 2008;7:1824–1837. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
