

Mutations in SDCCAG8/NPHP10 Cause Bardet-Biedl Syndrome and Are Associated with Penetrant Renal Disease and Absent Polydactyly
- PMID: 22190896
- PMCID: PMC3214956
- DOI: 10.1159/000331268
Mutations in SDCCAG8/NPHP10 Cause Bardet-Biedl Syndrome and Are Associated with Penetrant Renal Disease and Absent Polydactyly
Abstract
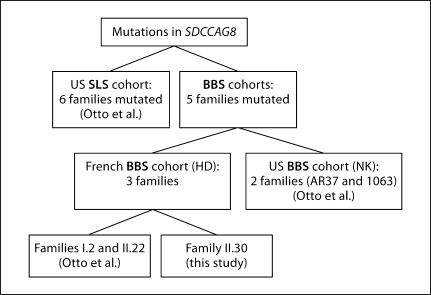
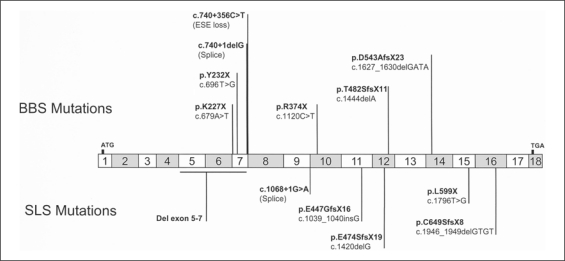
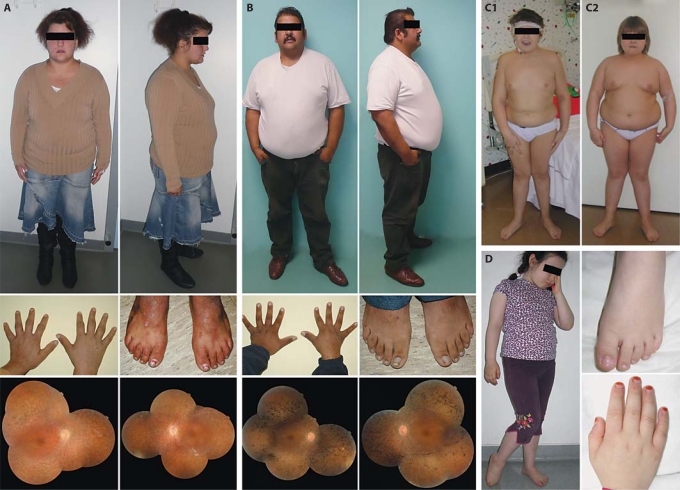
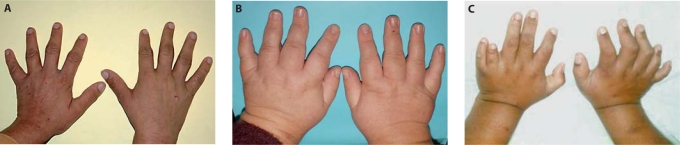
The ciliopathies are an expanding group of disorders caused by mutations in genes implicated in the biogenesis and function of primary cilia. Bardet-Biedl syndrome (BBS) is a model ciliopathy characterized by progressive retinal degeneration, obesity, polydactyly, cognitive impairment, kidney anomalies and hypogonadism. Mutations in SDCCAG8(NPHP10) were described recently in patients with nephronophthisis and retinal degeneration (Senior-Loken syndrome; SLS). Given the phenotypic and genetic overlap between known ciliopathy genes, we hypothesized that mutations in SDCCAG8 might also contribute alleles to more severe, multisystemic ciliopathies. We performed genetic and phenotypic analyses of 2 independent BBS cohorts. Subsequent to mutation screening, we made a detailed phenotypic analysis of 5 families mutated for SDCCAG8 (3 homozygous and 2 compound heterozygous mutations) and conducted statistical analyses across both cohorts to examine possible phenotype-genotype correlations with mutations at this locus. All patients with mutations in SDCCAG8 fulfilled the diagnostic criteria for BBS (retinal degeneration, obesity, cognitive defects, renal failure, hypogonadism). Interestingly, none of the patients with primary SDCCAG8 mutations had polydactyly, a frequent but not obligatory BBS feature. In contrast, the same patients displayed early-onset renal failure, obesity, as well as recurrent pulmonary and ENT infections. Comparison of the phenotypes of these families with our entire BBS cohort indicated that renal impairment and absent polydactyly correlated significantly with causal SDCCAG8 mutations. Thus, SDCCAG8 mutations are sufficient to cause BBS in 1-2% of our combined cohorts, and define this gene as the sixteenth BBS locus (BBS16). The absence of polydactyly and the concomitant, apparently fully penetrant association with early kidney failure represents the first significant genotype-phenotype correlation in BBS that potentially represents an indicator for phenotype-driven priority screening and informs specific patient management.
Figures
References
-
- Alström CH, Hallgren B, Nilsson LB, Asander H. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence-Moon-Bardet-Biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree. Acta Psychiatr Neurol Scand Suppl. 1959;129:1–35. - PubMed
-
- Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425:628–633. - PubMed
-
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–148. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
