

MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects
- PMID: 22192575
- PMCID: PMC3280279
- DOI: 10.1186/1471-2105-12-491
MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects
Abstract
Background: Second-generation sequencing technologies are precipitating major shifts with regards to what kinds of genomes are being sequenced and how they are annotated. While the first generation of genome projects focused on well-studied model organisms, many of today's projects involve exotic organisms whose genomes are largely terra incognita. This complicates their annotation, because unlike first-generation projects, there are no pre-existing 'gold-standard' gene-models with which to train gene-finders. Improvements in genome assembly and the wide availability of mRNA-seq data are also creating opportunities to update and re-annotate previously published genome annotations. Today's genome projects are thus in need of new genome annotation tools that can meet the challenges and opportunities presented by second-generation sequencing technologies.
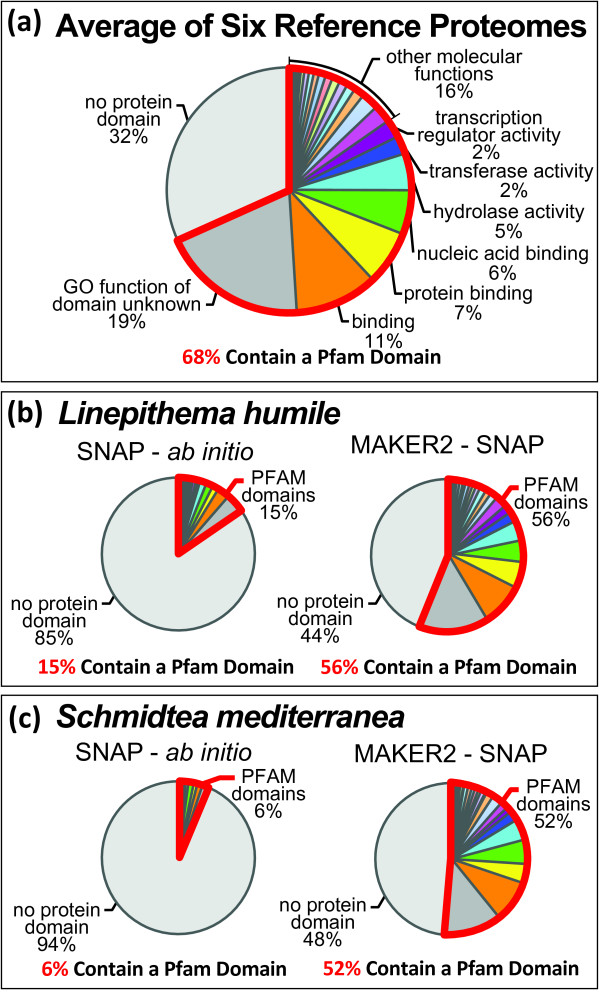
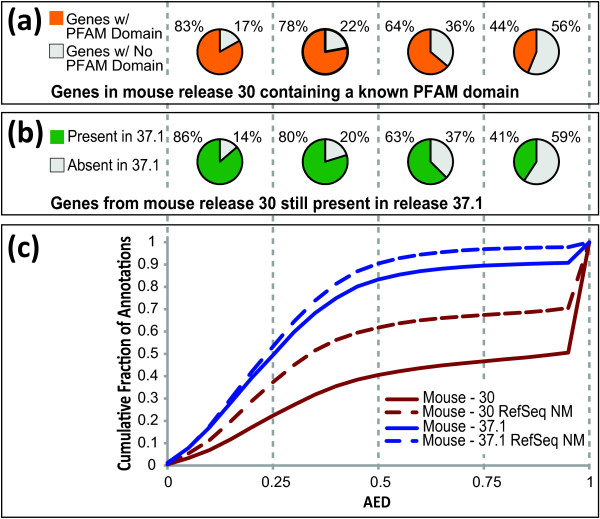
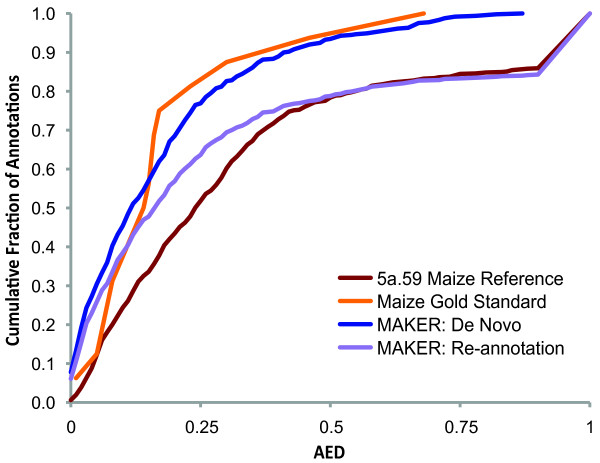
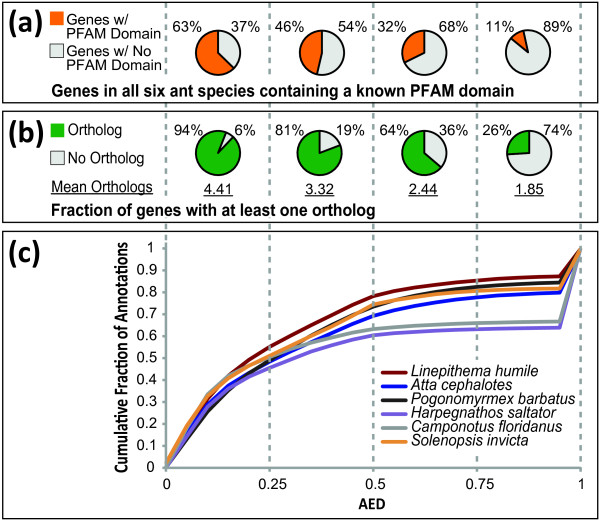
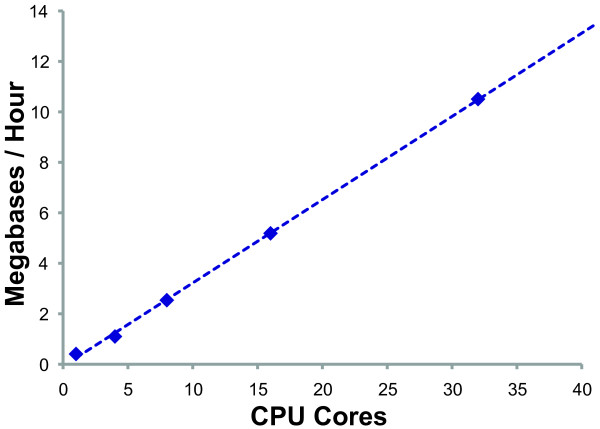
Results: We present MAKER2, a genome annotation and data management tool designed for second-generation genome projects. MAKER2 is a multi-threaded, parallelized application that can process second-generation datasets of virtually any size. We show that MAKER2 can produce accurate annotations for novel genomes where training-data are limited, of low quality or even non-existent. MAKER2 also provides an easy means to use mRNA-seq data to improve annotation quality; and it can use these data to update legacy annotations, significantly improving their quality. We also show that MAKER2 can evaluate the quality of genome annotations, and identify and prioritize problematic annotations for manual review.
Conclusions: MAKER2 is the first annotation engine specifically designed for second-generation genome projects. MAKER2 scales to datasets of any size, requires little in the way of training data, and can use mRNA-seq data to improve annotation quality. It can also update and manage legacy genome annotation datasets.
Figures
References
-
- The C. elegans Sequencing Consortium. Genome Sequence of the Nematode C. elegans: A Platform for Investigating Biology. Science. 1998;282(5396):2012–2018. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
