

Partner-aware prediction of interacting residues in protein-protein complexes from sequence data
- PMID: 22194998
- PMCID: PMC3237601
- DOI: 10.1371/journal.pone.0029104
Partner-aware prediction of interacting residues in protein-protein complexes from sequence data
Abstract
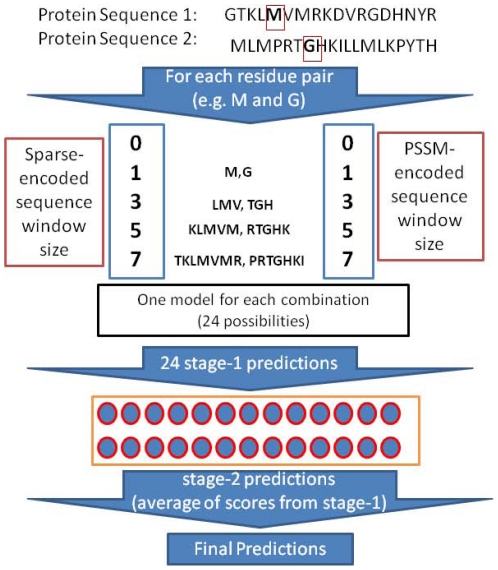
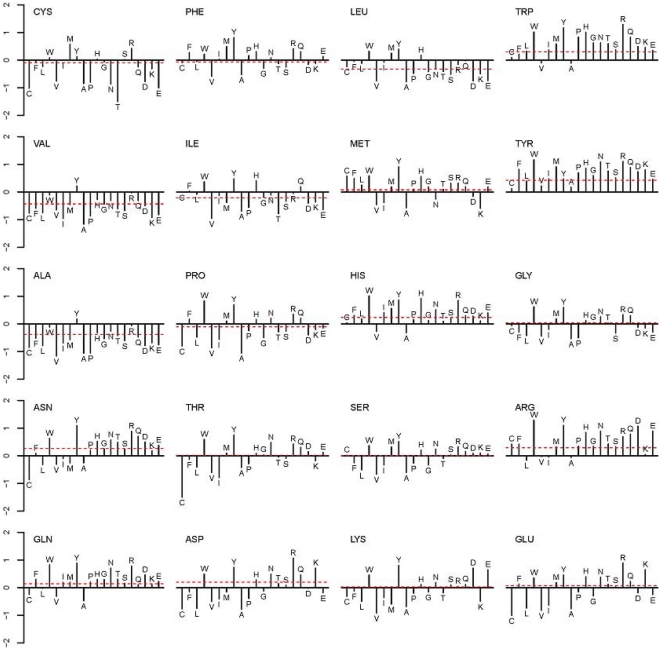
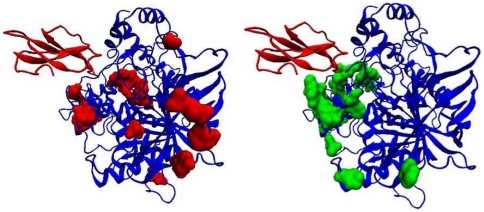
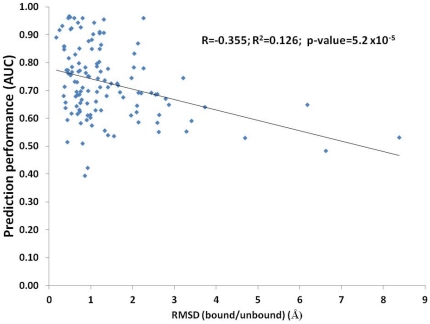
Computational prediction of residues that participate in protein-protein interactions is a difficult task, and state of the art methods have shown only limited success in this arena. One possible problem with these methods is that they try to predict interacting residues without incorporating information about the partner protein, although it is unclear how much partner information could enhance prediction performance. To address this issue, the two following comparisons are of crucial significance: (a) comparison between the predictability of inter-protein residue pairs, i.e., predicting exactly which residue pairs interact with each other given two protein sequences; this can be achieved by either combining conventional single-protein predictions or making predictions using a new model trained directly on the residue pairs, and the performance of these two approaches may be compared: (b) comparison between the predictability of the interacting residues in a single protein (irrespective of the partner residue or protein) from conventional methods and predictions converted from the pair-wise trained model. Using these two streams of training and validation procedures and employing similar two-stage neural networks, we showed that the models trained on pair-wise contacts outperformed the partner-unaware models in predicting both interacting pairs and interacting single-protein residues. Prediction performance decreased with the size of the conformational change upon complex formation; this trend is similar to docking, even though no structural information was used in our prediction. An example application that predicts two partner-specific interfaces of a protein was shown to be effective, highlighting the potential of the proposed approach. Finally, a preliminary attempt was made to score docking decoy poses using prediction of interacting residue pairs; this analysis produced an encouraging result.
Conflict of interest statement
Figures

References
-
- Hakes L, Pinney J, Robertson D, Lovell S. Protein-protein interaction networks and biology–what's the connection? Nature Biotechnology. 2008:69–72. - PubMed
-
- D'Alessandro A, Righetti P, Zolla L. The red blood cell proteome and interactome: an update. J Proteome Res. 2010;9:144–163. - PubMed
-
- Kelly W, Stumpf M. Protein-protein interactions: from global to local analyses. Curr Opin Biotechnol. 2008;19:396–403. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
