

The molecular basis of pharmacological chaperoning in human α-galactosidase
- PMID: 22195554
- PMCID: PMC3246215
- DOI: 10.1016/j.chembiol.2011.10.012
The molecular basis of pharmacological chaperoning in human α-galactosidase
Abstract
Fabry disease patients show a deficiency in the activity of the lysosomal enzyme α-galactosidase (α-GAL or α-Gal A). One proposed treatment for Fabry disease is pharmacological chaperone therapy, where a small molecule stabilizes the α-GAL protein, leading to increased enzymatic activity. Using enzyme kinetics, tryptophan fluorescence, circular dichroism, and proteolysis assays, we show that the pharmacological chaperones 1-deoxygalactonojirimycin (DGJ) and galactose stabilize the human α-GAL glycoprotein. Crystal structures of complexes of α-GAL and chaperones explain the molecular basis for the higher potency of DGJ over galactose. Using site-directed mutagenesis, we show the higher potency of DGJ results from an ionic interaction with D170. We propose that protonation of D170 in acidic conditions leads to weaker binding of DGJ. The results establish a biochemical basis for pharmacological chaperone therapy applicable to other protein misfolding diseases.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
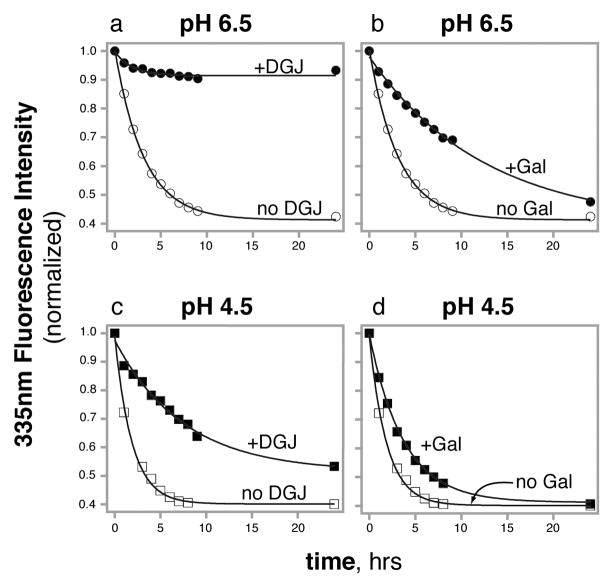
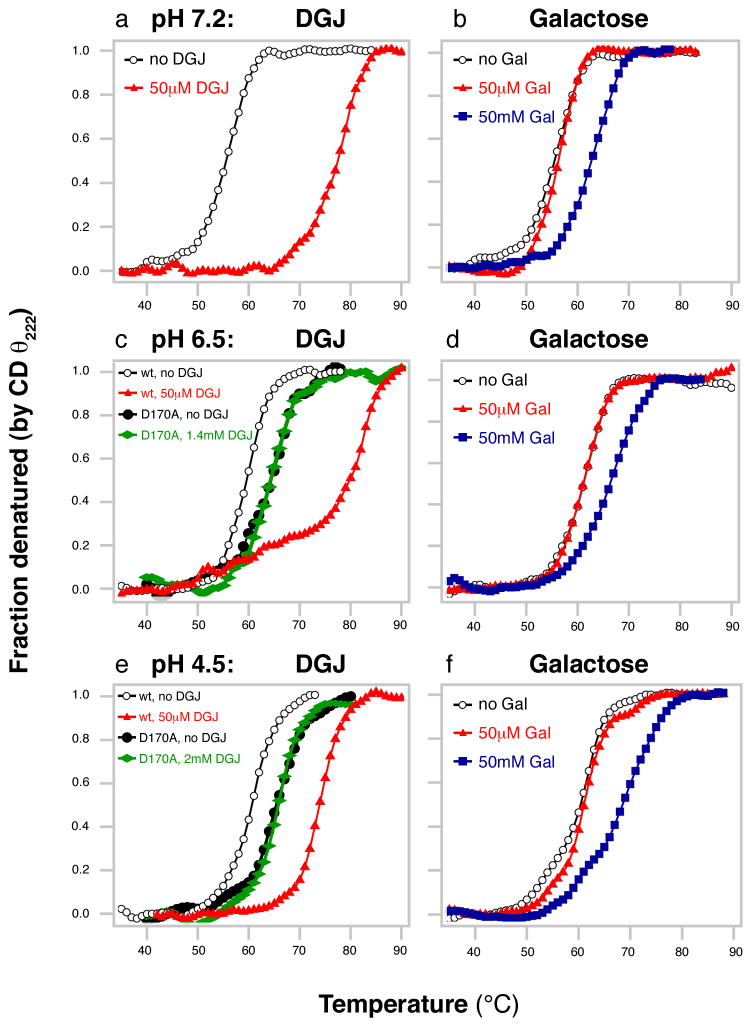
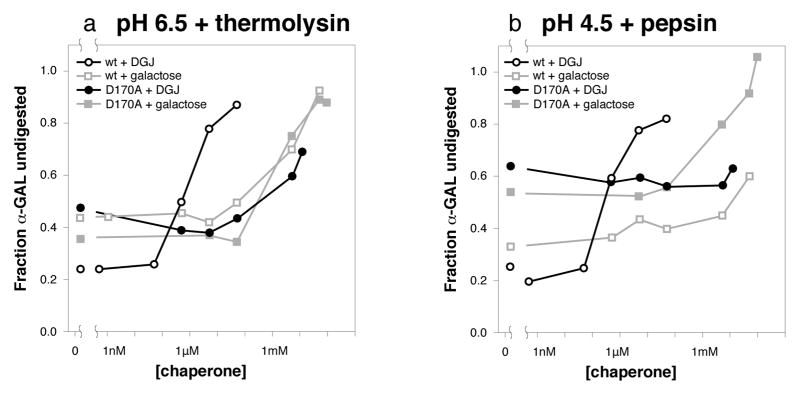
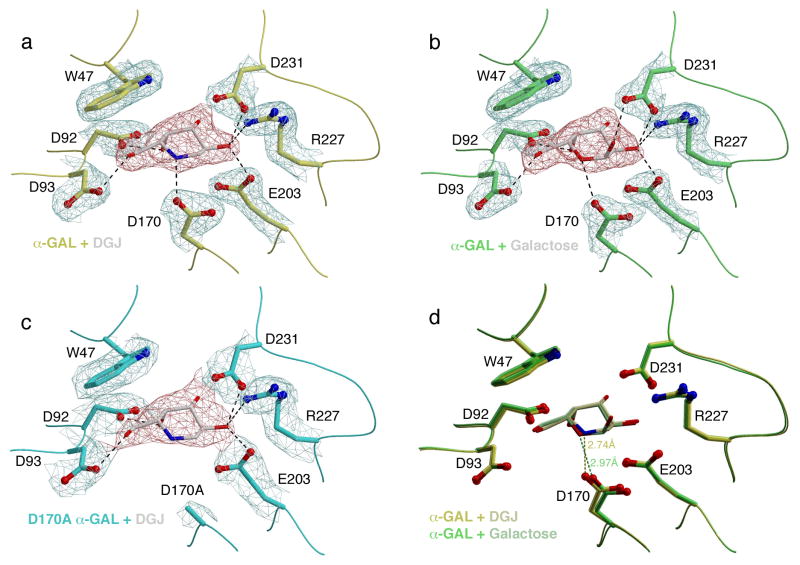
References
-
- Asano N, Ishii S, Kizu H, Ikeda K, Yasuda K, Kato A, Martin OR, Fan JQ. In vitro inhibition and intracellular enhancement of lysosomal α-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. Eur J Biochem. 2000;267:4179–4186. - PubMed
-
- Beutler E. Lysosomal storage diseases: natural history and ethical and economic aspects. Mol Genet Metab. 2006;88:208–215. - PubMed
-
- Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967;276:1163–1167. - PubMed
-
- Cohen FE, Kelly JW. Therapeutic approaches to protein-misfolding diseases. Nature. 2003;426:905–909. - PubMed
-
- Desnick RJ, Ioannou YA, Eng CM. α-Galactosidase A Deficiency: Fabry Disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 3733–3774.
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
