

Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome
- PMID: 22197486
- PMCID: PMC3257878
- DOI: 10.1016/j.ajhg.2011.11.021
Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome
Abstract
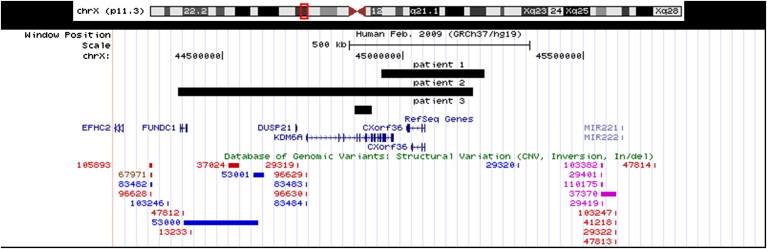
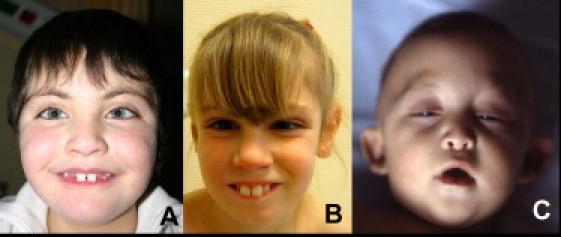
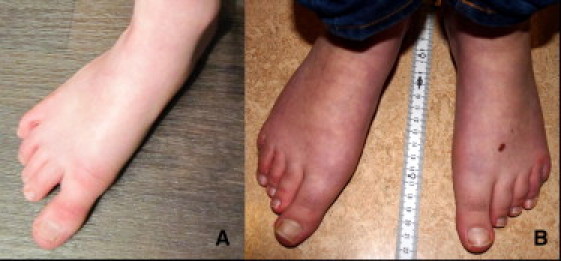
Kabuki syndrome (KS) is a rare genetic disease that causes developmental delay and congenital anomalies. Since the identification of MLL2 mutations as the primary cause of KS, such mutations have been identified in 56%-76% of affected individuals, suggesting that there may be additional genes associated with KS. Here, we describe three KS individuals with de novo partial or complete deletions of an X chromosome gene, KDM6A, that encodes a histone demethylase that interacts with MLL2. Although KDM6A escapes X inactivation, we found a skewed X inactivation pattern, in which the deleted X chromosome was inactivated in the majority of the cells. This study identifies KDM6A mutations as another cause of KS and highlights the growing role of histone methylases and histone demethylases in multiple-congenital-anomaly and intellectual-disability syndromes.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Niikawa N., Matsuura N., Fukushima Y., Ohsawa T., Kajii T. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J. Pediatr. 1981;99:565–569. - PubMed
-
- Kuroki Y., Suzuki Y., Chyo H., Hata A., Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J. Pediatr. 1981;99:570–573. - PubMed
-
- Schoumans J., Nordgren A., Ruivenkamp C., Brøndum-Nielsen K., Teh B.T., Annéren G., Holmberg E., Nordenskjöld M., Anderlid B.M. Genome-wide screening using array-CGH does not reveal microdeletions/microduplications in children with Kabuki syndrome. Eur. J. Hum. Genet. 2005;13:260–263. - PubMed
-
- Miyake N., Shimokawa O., Harada N., Sosonkina N., Okubo A., Kawara H., Okamoto N., Ohashi H., Kurosawa K., Naritomi K., et al. No detectable genomic aberrations by BAC array CGH in Kabuki make-up syndrome patients. Am. J. Med. Genet. A. 2006;140:291–293. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
