

Exome sequence identifies RIPK4 as the Bartsocas-Papas syndrome locus
- PMID: 22197488
- PMCID: PMC3257958
- DOI: 10.1016/j.ajhg.2011.11.013
Exome sequence identifies RIPK4 as the Bartsocas-Papas syndrome locus
Abstract
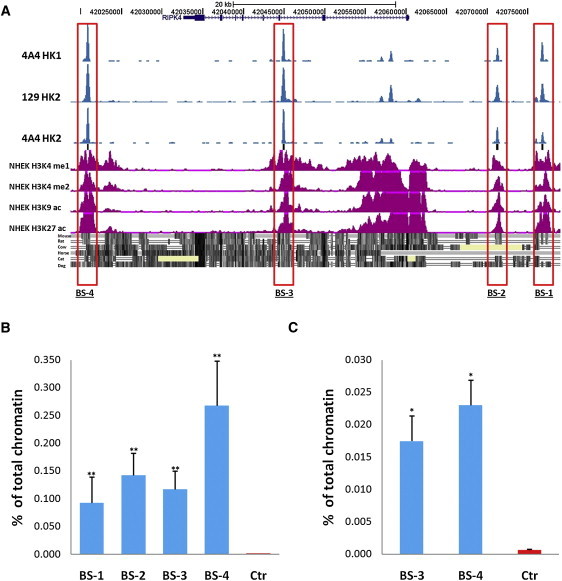
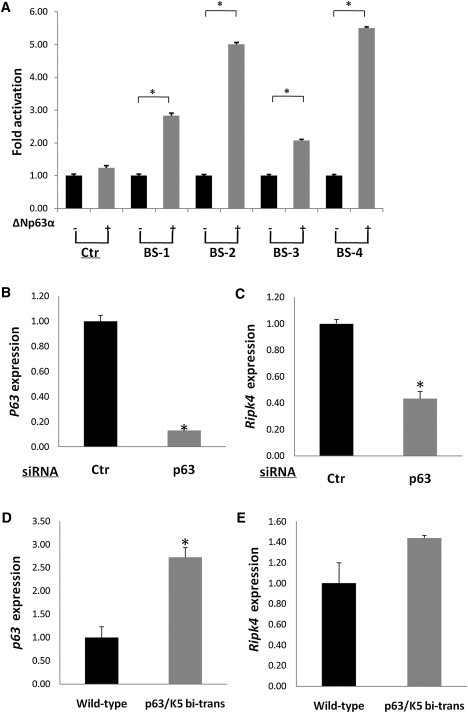
Pterygium syndromes are complex congenital disorders that encompass several distinct clinical conditions characterized by multiple skin webs affecting the flexural surfaces often accompanied by craniofacial anomalies. In severe forms, such as in the autosomal-recessive Bartsocas-Papas syndrome, early lethality is common, complicating the identification of causative mutations. Using exome sequencing in a consanguineous family, we identified the homozygous mutation c.1127C>A in exon 7 of RIPK4 that resulted in the introduction of the nonsense mutation p.Ser376X into the encoded ankyrin repeat-containing kinase, a protein that is essential for keratinocyte differentiation. Subsequently, we identified a second mutation in exon 2 of RIPK4 (c.242T>A) that resulted in the missense variant p.Ile81Asn in the kinase domain of the protein. We have further demonstrated that RIPK4 is a direct transcriptional target of the protein p63, a master regulator of stratified epithelial development, which acts as a nodal point in the cascade of molecular events that prevent pterygium syndromes.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Fuchs E., Raghavan S. Getting under the skin of epidermal morphogenesis. Nat. Rev. Genet. 2002;3:199–209. - PubMed
-
- Watt F.M. Stem cell fate and patterning in mammalian epidermis. Curr. Opin. Genet. Dev. 2001;11:410–417. - PubMed
-
- Lahtela J., Nousiainen H.O., Stefanovic V., Tallila J., Viskari H., Karikoski R., Gentile M., Saloranta C., Varilo T., Salonen R., et al. Mutant CHUK and severe fetal encasement malformation. N. Engl. J. Med. 2010;363:1631–1637. - PubMed
-
- Veenstra-Knol H.E., Kleibeuker A., Timmer A., ten Kate L.P., van Essen A.J. Unreported manifestations in two Dutch families with Bartsocas-Papas syndrome. Am. J. Med. Genet. 2003;123A:243–248. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
