

Database independent proteomics analysis of the ostrich and human proteome
- PMID: 22198768
- PMCID: PMC3258627
- DOI: 10.1073/pnas.1108399108
Database independent proteomics analysis of the ostrich and human proteome
Abstract
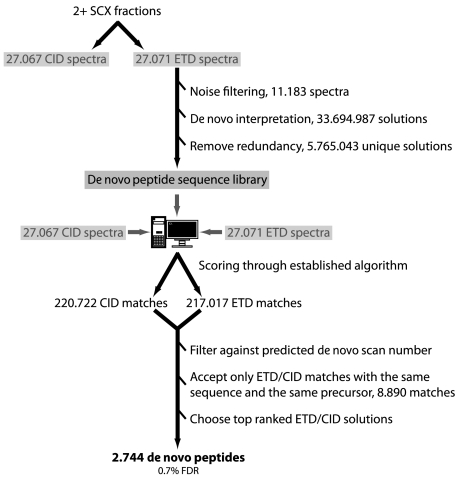
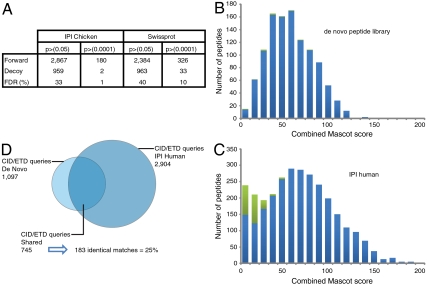
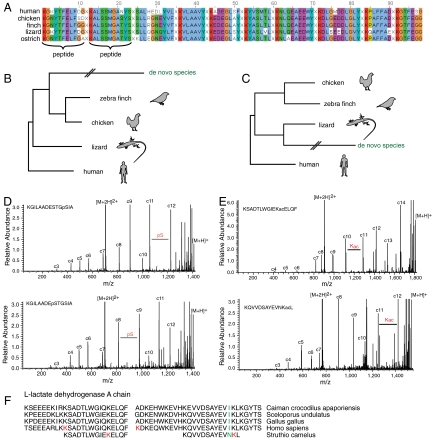
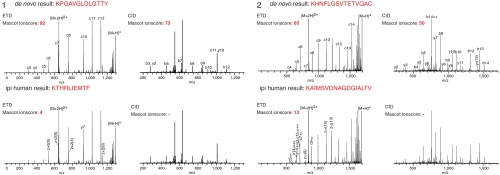
Mass spectrometry (MS)-based proteome analysis relies heavily on the presence of complete protein databases. Such a strategy is extremely powerful, albeit not adequate in the analysis of unpredicted postgenome events, such as posttranslational modifications, which exponentially increase the search space. Therefore, it is of interest to explore "database-free" approaches. Here, we sampled the ostrich and human proteomes with a method facilitating de novo sequencing, utilizing the protease Lys-N in combination with electron transfer dissociation. By implementing several validation steps, including the combined use of collision-induced dissociation/electron transfer dissociation data and a cross-validation with conventional database search strategies, we identified approximately 2,500 unique de novo peptide sequences from the ostrich sample with over 900 peptides generating full backbone sequence coverage. This dataset allowed the appropriate positioning of ostrich in the evolutionary tree. The described database-free sequencing approach is generically applicable and has great potential in important proteomics applications such as in the analysis of variable parts of endogenous antibodies or proteins modified by a plethora of complex posttranslational modifications.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Exploring new proteome space: combining Lys-N proteolytic digestion and strong cation exchange (SCX) separation in peptide-centric MS-driven proteomics.Methods Mol Biol. 2011;753:157-67. doi: 10.1007/978-1-61779-148-2_11. Methods Mol Biol. 2011. PMID: 21604122
-
Proteomics-grade de novo sequencing approach.J Proteome Res. 2005 Nov-Dec;4(6):2348-54. doi: 10.1021/pr050288x. J Proteome Res. 2005. PMID: 16335984
-
Extended Range Proteomic Analysis (ERPA): a new and sensitive LC-MS platform for high sequence coverage of complex proteins with extensive post-translational modifications-comprehensive analysis of beta-casein and epidermal growth factor receptor (EGFR).J Proteome Res. 2005 Jul-Aug;4(4):1155-70. doi: 10.1021/pr050113n. J Proteome Res. 2005. PMID: 16083266
-
Proteomics by mass spectrometry--go big or go home?J Pharm Biomed Anal. 2011 Jun 25;55(4):832-41. doi: 10.1016/j.jpba.2011.02.012. Epub 2011 Feb 17. J Pharm Biomed Anal. 2011. PMID: 21382686 Review.
-
Classification and identification of bacteria using mass spectrometry-based proteomics.Expert Rev Proteomics. 2005 Dec;2(6):863-78. doi: 10.1586/14789450.2.6.863. Expert Rev Proteomics. 2005. PMID: 16307516 Review.
Cited by
-
Neutron-encoded signatures enable product ion annotation from tandem mass spectra.Mol Cell Proteomics. 2013 Dec;12(12):3812-23. doi: 10.1074/mcp.M113.028951. Epub 2013 Sep 16. Mol Cell Proteomics. 2013. PMID: 24043425 Free PMC article.
-
Innovative in Silico Approaches for Characterization of Genes and Proteins.Front Genet. 2022 May 18;13:865182. doi: 10.3389/fgene.2022.865182. eCollection 2022. Front Genet. 2022. PMID: 35664302 Free PMC article. Review.
-
Expanding the detectable HLA peptide repertoire using electron-transfer/higher-energy collision dissociation (EThcD).Proc Natl Acad Sci U S A. 2014 Mar 25;111(12):4507-12. doi: 10.1073/pnas.1321458111. Epub 2014 Mar 10. Proc Natl Acad Sci U S A. 2014. PMID: 24616531 Free PMC article.
-
Next-generation proteomics: towards an integrative view of proteome dynamics.Nat Rev Genet. 2013 Jan;14(1):35-48. doi: 10.1038/nrg3356. Epub 2012 Dec 4. Nat Rev Genet. 2013. PMID: 23207911 Review.
References
-
- McLafferty FW. A century of progress in molecular mass spectrometry. Annu Rev Anal Chem. 2011;4:1–22. - PubMed
-
- Cox J, Mann M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu Rev Biochem. 2011;80:273–299. - PubMed
-
- Sadygov RG, Cociorva D, Yates JR., 3rd Large-scale database searching using tandem mass spectra: Looking up the answer in the back of the book. Nat Methods. 2004;1:195–202. - PubMed
-
- Pappin DJ, Hojrup P, Bleasby AJ. Rapid identification of proteins by peptide-mass fingerprinting. Curr Biol. 1993;3:327–332. - PubMed
-
- Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. A nonergodic process. J Am Chem Soc. 1998;120:3265–3266.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
