

Protein aggregates in Huntington's disease
- PMID: 22200539
- PMCID: PMC3909772
- DOI: 10.1016/j.expneurol.2011.12.013
Protein aggregates in Huntington's disease
Abstract
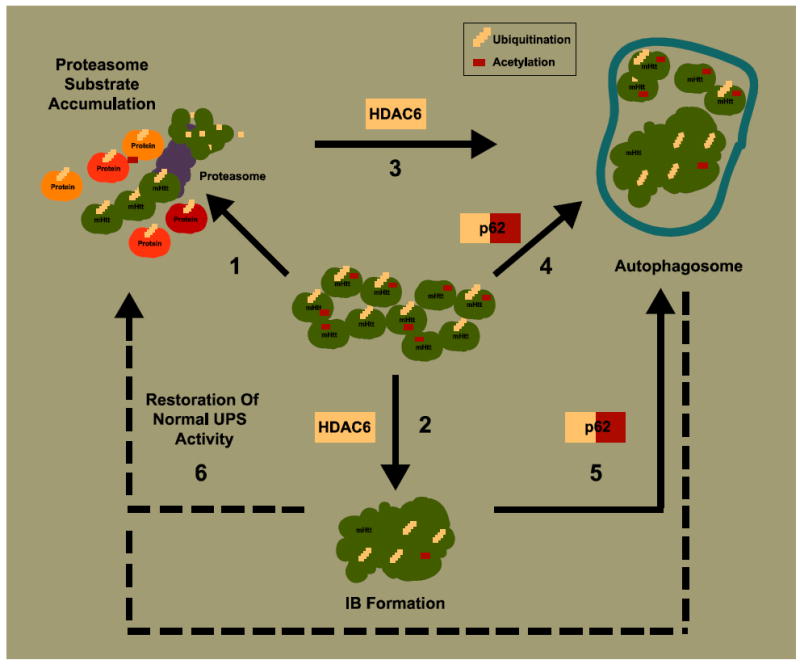
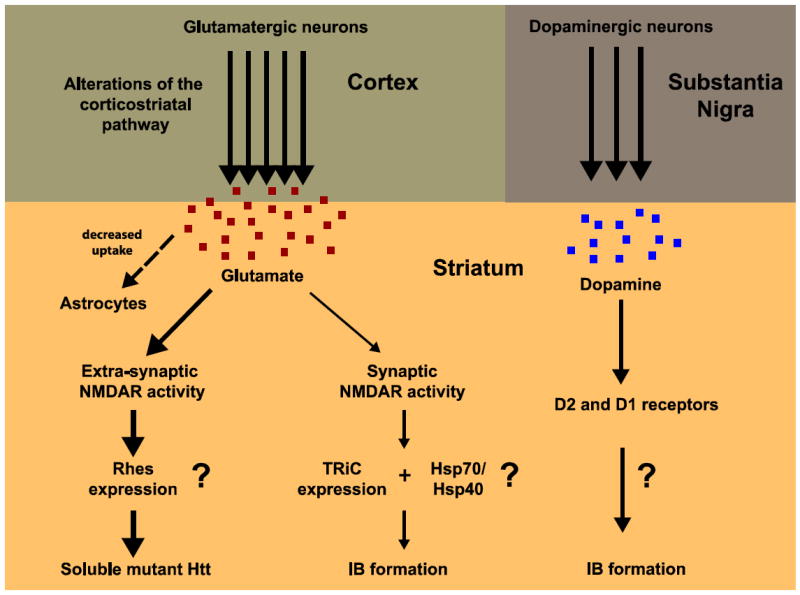
Huntington's disease (HD) is an incurable neurodegenerative disease characterized by abnormal motor movements, personality changes, and early death. HD is caused by a mutation in the IT-15 gene that expands abnormally the number of CAG nucleotide repeats. As a result, the translated protein huntingtin contains disease-causing expansions of glutamines (polyQ) that make it prone to misfold and aggregate. While the gene and mutations that cause HD are known, the mechanisms underlying HD pathogenesis are not. Here we will review the state of knowledge of HD, focusing especially on a hallmark pathological feature-intracellular aggregates of mutant Htt called inclusion bodies (IBs). We will describe the role of IBs in the disease. We speculate that IB formation could be just one component of a broader coping response triggered by misfolded Htt whose efficacy may depend on the extent to which it clears toxic forms of mutant Htt. We will describe how IB formation might be regulated and which factors could determine different coping responses in different subsets of neurons. A differential regulation of IB formation as a function of the cellular context could, eventually, explain part of the neuronal vulnerability observed in HD.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
References
-
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. - PubMed
-
- Atwal RS, Desmond CR, Caron N, Maiuri T, Xia J, Sipione S, Truant R. Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat Chem Biol. 2011;7:453–460. - PubMed
-
- Becher MW, Kotzuk JA, Sharp AH, Davies SW, Bates GP, Price DL, Ross CA. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis. 1998;4:387–397. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
