

Kinetic studies of the reactions of O(2) and NO with reduced Thermus thermophilus ba(3) and bovine aa(3) using photolabile carriers
- PMID: 22201543
- PMCID: PMC3498500
- DOI: 10.1016/j.bbabio.2011.12.005
Kinetic studies of the reactions of O(2) and NO with reduced Thermus thermophilus ba(3) and bovine aa(3) using photolabile carriers
Abstract
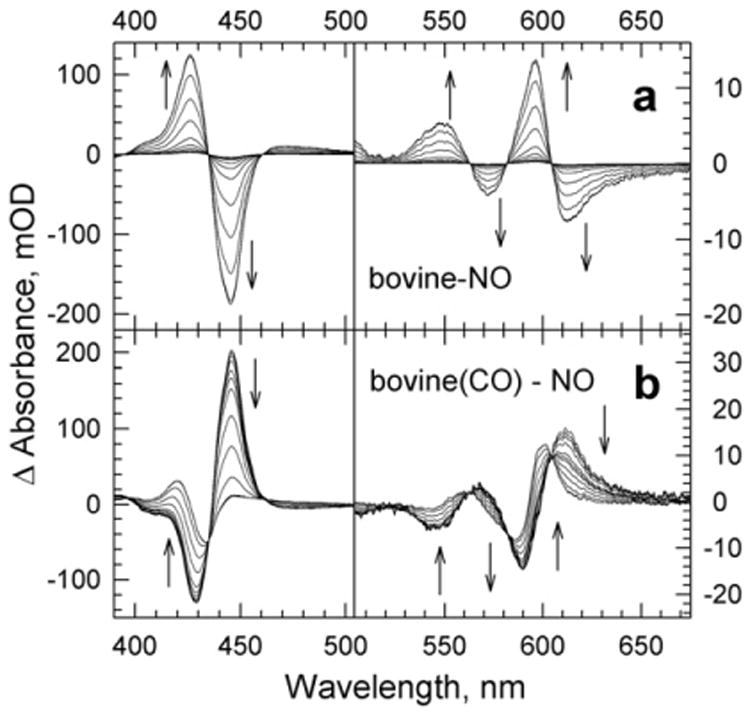
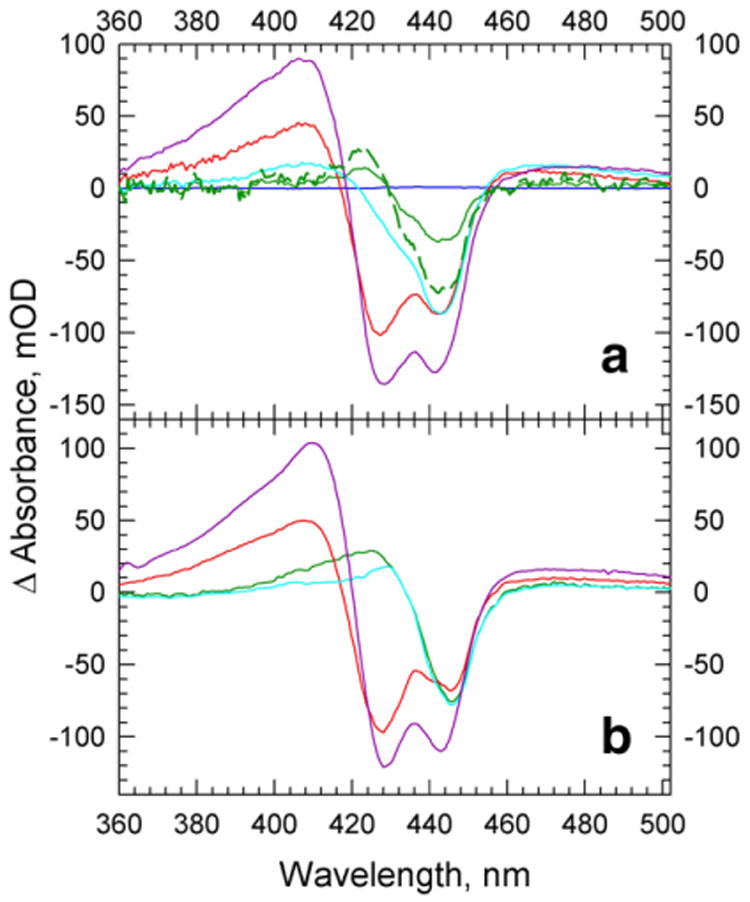
The reactions of molecular oxygen (O(2)) and nitric oxide (NO) with reduced Thermus thermophilus (Tt) ba(3) and bovine heart aa(3) were investigated by time-resolved optical absorption spectroscopy to establish possible relationships between the structural diversity of these enzymes and their reaction dynamics. To determine whether the photodissociated carbon monoxide (CO) in the CO flow-flash experiment affects the ligand binding dynamics, we monitored the reactions in the absence and presence of CO using photolabile O(2) and NO complexes. The binding of O(2)/NO to reduced ba(3) in the absence of CO occurs with a second-order rate constant of 1×10(9)M(-1)s(-1). This rate is 10-times faster than for the mammalian enzyme, and which is attributed to structural differences in the ligand channels of the two enzymes. Moreover, the O(2)/NO binding in ba(3) is 10-times slower in the presence of the photodissociated CO while the rates are the same for the bovine enzyme. This indicates that the photodissociated CO directly or indirectly impedes O(2) and NO access to the active site in Tt ba(3), and that traditional CO flow-flash experiments do not accurately reflect the O(2) and NO binding kinetics in ba(3). We suggest that in ba(3) the binding of O(2) (NO) to heme a(3)(2+) causes rapid dissociation of CO from Cu(B)(+) through steric or electronic effects or, alternatively, that the photodissociated CO does not bind to Cu(B)(+). These findings indicate that structural differences between Tt ba(3) and the bovine aa(3) enzyme are tightly linked to mechanistic differences in the functions of these enzymes. This article is part of a Special Issue entitled: Respiratory Oxidases.
Copyright © 2011 Elsevier B.V. All rights reserved.
Figures






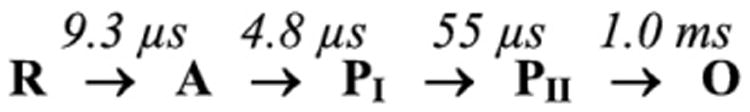
References
-
- Brzezinski P, Larsson G. Redox-driven proton pumping by heme–copper oxidases. Biochim Biophys Acta. 2003;1605:1–13. - PubMed
-
- Ferguson-Miller S, Babcock GT. Heme/copper terminal oxidases. Chem Rev. 1996;96:2889–2907. - PubMed
-
- Kaila VR, Verkhovsky MI, Wikström M. Proton-coupled electron transfer in cytochrome oxidase. Chem Rev. 2010;110:7062–7081. - PubMed
-
- Einarsdóttir Ó. Fast reactions of cytochrome oxidase. Biochim Biophys Acta. 1995;1229:129–147. - PubMed
-
- Wikström MKF. Proton pump coupled to cytochrome c oxidase in mitochondria. Nature. 1977;266:271–273. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
